Neurofibromatosis tipo I. Etiología, diagnóstico y tratamiento
Fernanda Deloya-Ardón María ORCID: 0009-0003-5811-8212
Axel García-Burgo ORCID: 0000-0001-7362-3928
CIENCIA UANL / AÑO 27, No.123, enero-febrero 2024
DOI: https://doi.org/10.29105/cienciauanl27.123-4
RESUMEN
La neurofibromatosis tipo I es una enfermedad neurodermatológica con herencia autosómico dominante, relacionada con el gen NF1. Es considerada “una enfermedad genética, de herencia autosómica dominante, de alta penetrancia y expresividad variable” (Galán, 2014). Sus manifestaciones pueden ser numerosas y las afectaciones son mayoritariamente en piel, máculas cafés con leche, en el sistema nervioso periférico y central (García, Cervini y Pierini, 2013), alteraciones en zona de la columna y lumbar. Su diagnóstico se basa en las manifestaciones clínicas y en los estudios por imagen, por lo cual debe tener un enfoque multidisciplinario, con especialistas en pediatría, ortopedia, dermatología y neurología, debido a las lesiones que engloba en los pacientes, por lo general niños, en la función de la marcha y a nivel de la piel.
Palabras clave: neurofibromatosis, genética, autonómica, periférico y central.
ABSTRACT
Keywords: neurofibromatosis, genetic, autosomal, peripheral, central.
La neurofibromatosis tipo I se conocía como enfermedad de von Recklinghausen, una genodermatosis cuya principal característica son máculas o manchas color café con leche y presencia de neurofibromas (Salas-Alanís, De la Garza-Ramos y Cepeda-Valdés, 2011). Existen siete variaciones: clásica o von Recklinghausen, central, mixta, variante, segmentaria, con manchas sin neurofibromas y de inicio tardío. Este padecimiento, con sus variantes, tiene un antecedente ya abordado por Friederich von Recklinghausen en 1882, quien observó pequeñas y medianas manchas de color café en extremidades superiores e inferiores.
En ésta existe un factor que influye o determina el comienzo y la aparición de las manifestaciones clínicas. Es ocasionada por el gen localizado en el cromosoma 17, éste posee un componente llamado neurofibromina, relacionado con la diferenciación y formación de tejidos celulares: dermatológicos y nerviosos (Jiménez-Caballero, 2013). Asimismo, posee gran relevancia en los protooncogenes, llamados ras, por tanto, si existe alguna disfunción en éste componente genético, habría relación con la formación de tumores que podrían ser malignos (astrocitomas) o benignos (neurofibromas plexiformes), además de distintas manifestaciones en la presión arterial (Ríos-Sanabria y Mora-Hernández, 2014).
EPIDEMIOLOGÍA
Se ha observado que afecta a uno de cada 3,000 individuos, en uno de cada 200 con retraso mental, 50% de los casos presentan neomutaciones, a partir de este punto representa un reto al momento del diagnóstico, debido a que por su alta penetrancia y dominancia surgen diversas manifestaciones clínicas y se debe tomar en cuenta tanto la historia natural de la enfermedad, un estudio prospectivo a lo largo de la vida del individuo y la aparición de tumores benignos y malignos.
SIGNOS Y SÍNTOMAS DE LA NEUROFIBROMATOSIS TIPO I
En diversas guías, artículos y sesiones mencionadas, se describen múltiples signos y síntomas: “retraso mental, cefaleas, convulsiones, parálisis, neurofibromas, glaucomas, disminución de la agudeza visual, baja estatura, desviación de la columna (escoliosis), constipación, obstrucción intestinal y tumores del sistema nervioso central” (Gómez y Batista, 2015).
MANIFESTACIONES DERMATOLÓGICAS
El Instituto Nacional de la Salud de EE UU definió, en 1987, los criterios de la neurofibromatosis tipo I; para su diagnóstico debe tener al menos dos de siete, entre los que destacan: seis o más manchas cafés con leche, iguales o mayores de 5 mm en prepúberes y de 15 mm de diámetro en pospúberes. Dos o más neurofibromas de cualquier clase. Se describen efélides en la región inguinal o axilar (Llorente-La Orden et al., 2018). Las manifestaciones cutáneas presentan distintos patrones de crecimiento a lo largo de la edad, destacando entre los dos a seis años, aquí las efélides axilares, máculas cafés con leche, son los más característicos en extremidades superiores, dicha situación es detectada en el desarrollo del menor y los especialistas sugieren monitoreo continuo.
Figura 1. El patrón característico son las máculas cafés con leche, las cuales aparecen en extremidades superiores e inferiores, principalmente en pacientes pediátricos (antes de los 5 años) (Dermadol, 2014).
MANIFESTACIONES OFTALMOLÓGICAS
Dentro del desarrollo y la historia natural de la afección, los cambios evidentes son oftamológicos: nódulos de Lisch, es decir, tumores pigmentarios que a la exploración se consideran lesiones sobreelevadas, superficie lisa, de manera bilateral, el mismo patrón “café con leche”, no afectan la visión. “Los gliomas de vía óptica suelen presentarse normalmente antes de los 5 años. Aproximadamente entre 10 y 25% de los sujetos con estas lesiones tienen desenlaces poco alentadores: pérdida o disminución de la agudeza visual, alteración de la visión de colores, principalmente si se trata de localización en el quiasma óptico” (Domínguez-Hernández, 2014). Por esto es importante el seguimiento de la región anatómica y evaluación para valoración de tratamiento, dependiendo de la edad y condición física.
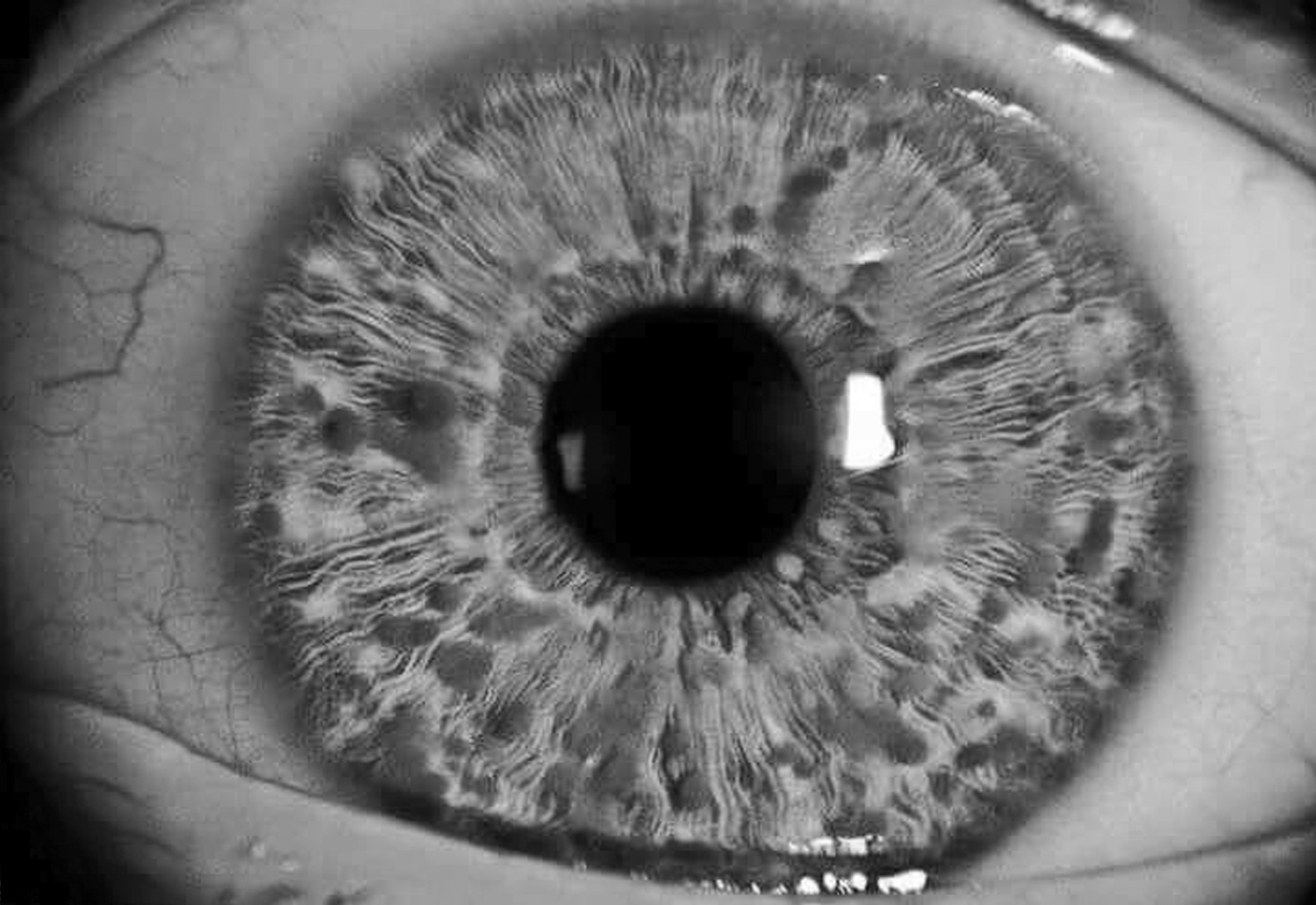
Figura 2. Los nódulos de Lisch consisten en hamartomas melanocíticos que aparecen en el iris, se asocian a neurofibromatosis tipo 1, también enfermedad de von Recklinghausen (Martínez, 2016).
MANIFESTACIONES NEUROLÓGICAS
Aparecen alteraciones de nivel neurológico, en el desenvolvimiento psicológico, social, motriz y de aprendizaje. En el deterioro hay disminución de la habilidad de razonamiento y la capacidad de pensamiento suele ser leve. En ocasiones hay dificultad de aprendizaje específica, problemas de lectura o redacción y lógico-matemática. “Suele presentarse el trastorno de déficit de atención por hiperactividad. Parte del cuadro neurológico en la neurofibromatosis tipo I está relacionado con la talla cefálica, que se reporta mayor al promedio ocasionado por el volumen cerebral. Simultáneamente, la persona afectada puede llegar a manifestar crisis convulsivas, compresión medular y déficit neurológico” (Pardo-Somalo y Málago-Guerrero, 2014), debido a tumores alojados en la estructura encefálica.
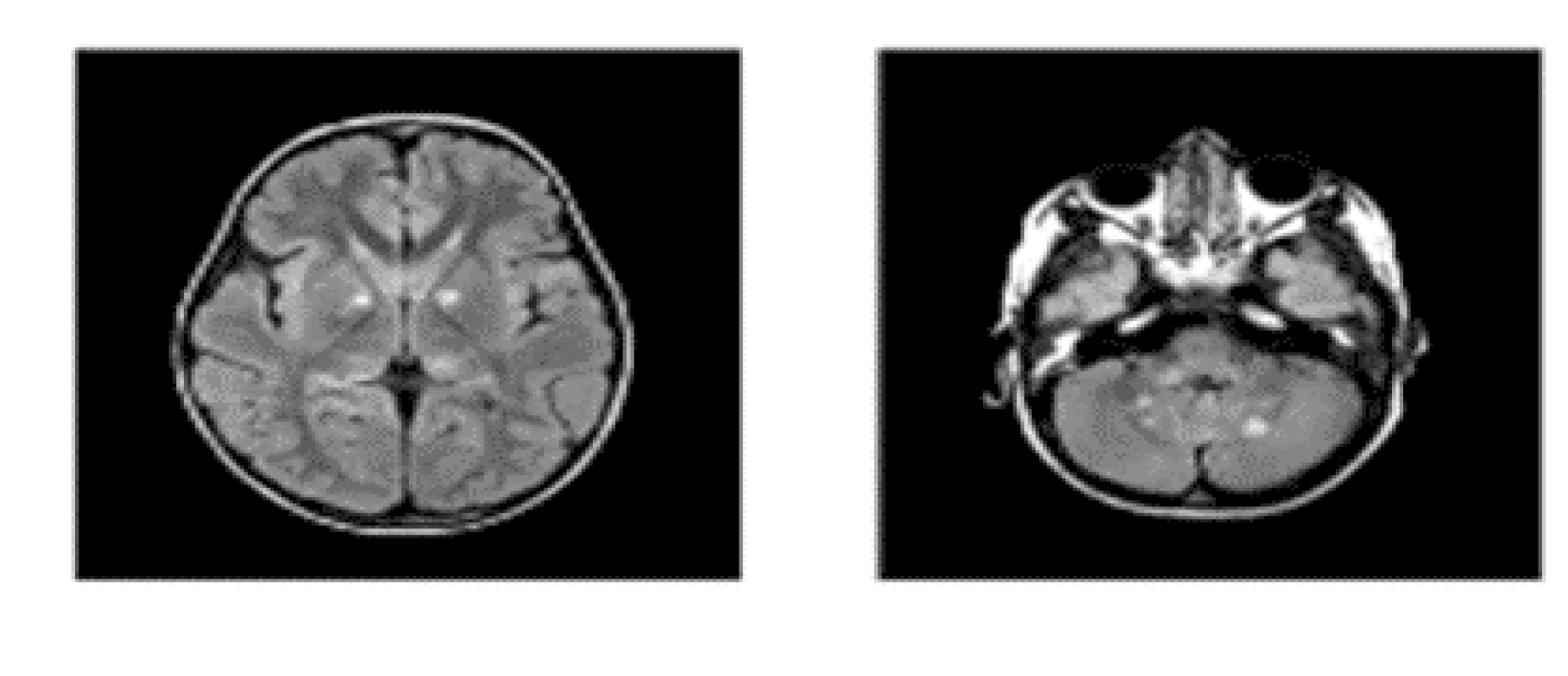
Figura 3. En estas imágenes se pueden observar alteraciones en la masa encefálica y macrocefalia (Neurofibromatosis, 2017).
MANIFESTACIONES TENSIONALES
Los efectos posteriores a la aparición de la neurofibromatosis pueden tener relación con la hipertensión arterial y con las feocromocitomas, principalmente en adultos, tales manifestaciones vienen acompañadas de “síndromes neurálgicos como cefalea tensional, migraña, fósfenos” (Bersusky, 2017). Por este motivo, el especialista debe realizar una inspección exhaustiva, en interrogatorio y en exploración física.
MANIFESTACIONES MUSCULOESQUELÉTICAS
Esta clase de alteraciones morfológicas ocurren en sitios anatómicos: cuerpos vertebrales, cuyo resultado son “lesiones distróficas, cifosis cervical; los pacientes reportan, al momento de la consulta y a la exploración, dolor cervical. La cifoescoliosis y lordoescoliosis torácica son un cuadro característico” (Gómez y Batista, 2015), resulta importante la detección de dicho cuadro, junto con las apariciones dérmicas (manchas café con leche), dificultad en aprendizaje y alteraciones en las cifras tensionales.
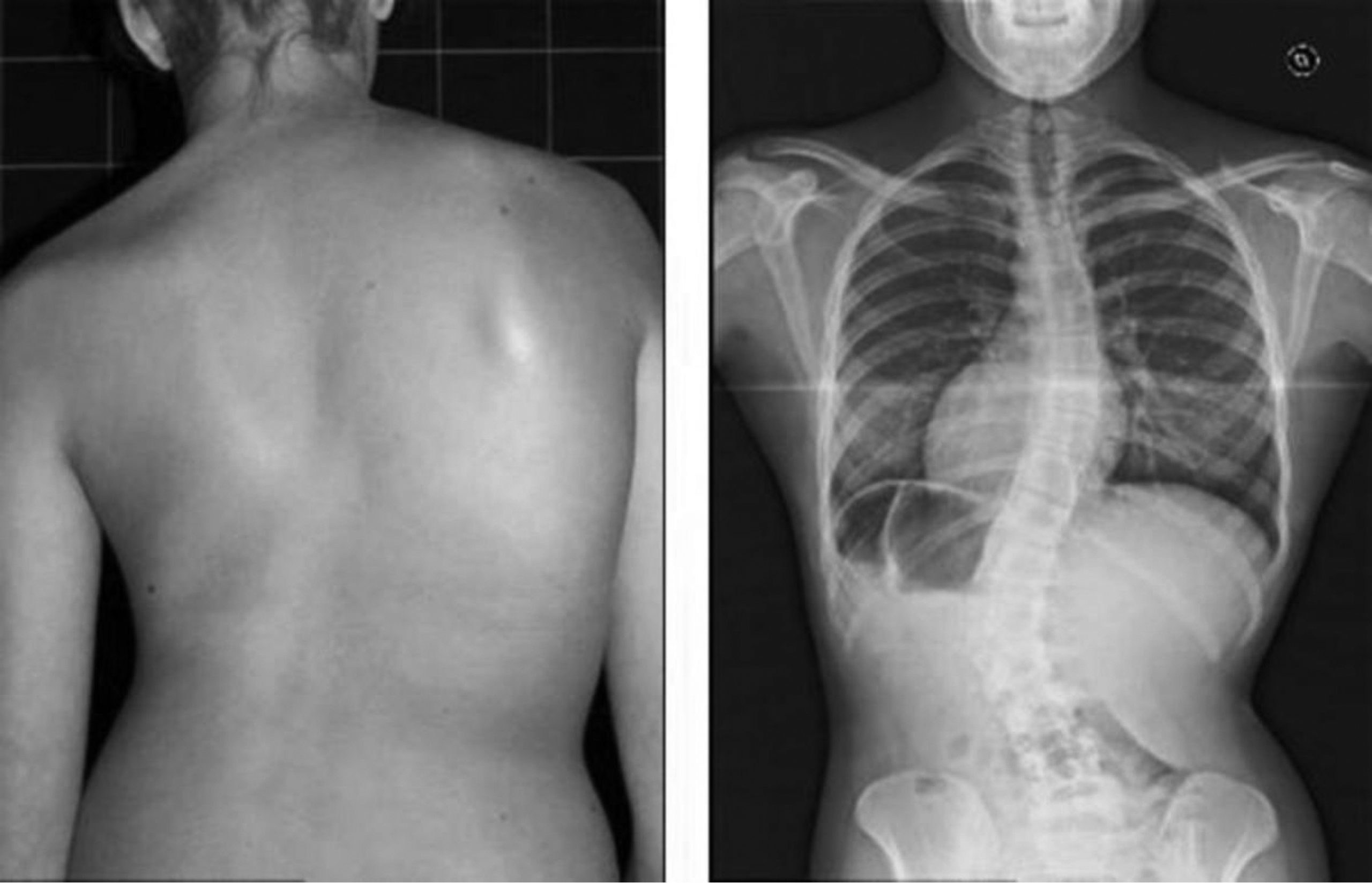
Figura 4. Se reportan en casos clínicos con alteraciones en la columna vertebral, dolor agudo y crónico, dependiendo de la gravedad del padecimiento, se pueden detectar con estudios de imagen como la radiografía de columna (Fisioterapia, 2016).
DIAGNÓSTICO DIFERENCIAL DE LA NEUROFIBROMATOSIS TIPO II
Pensando en diagnósticos diferenciales, la neurofibromatosis tipo II es una forma familiar: “manchas café con leche, síndrome LEOPARD (lentigos, anormalidades en el EKG, hipertelorismo ocular, estenosis ocular, genitales anormales, retraso anormal en el crecimiento y sordera neurosensorial)” (Salas-Alanís, De la Garza-Ramos y Cepeda-Valdés, 2011); existen otros dictámenes diferenciales, por ejemplo: el síndrome McCune-Albright (displasia fibrosa poliostótica), esclerosis tuberosa, feocromocitomas, carcinoma medular tiroidea, hiperplasia paratiroidea, neuromas mucocutáneos, las cuales tienen signos ysíntomas similares a la neurofibromatosis tipo II. No obstante, la diferencia principal es la existencia de componentes hormonales, afectando el desarrollo en pacientes menores de 5 años.
TRATAMIENTO EN LA NEUROFIBROMATOSIS TIPO I
El tratamiento principal consiste en una revisión especializada –por parte de quien lo trata– del sistema nervioso central y periférico, óseo, cardiovascular, oftalmológico y dermatológico. Específicamente los neurofibromas cutáneos o subcutáneos, por aspecto clínico y estético, deben ser removidos, dependiendo las características, morfología, tamaño y avance, puede determinarse, si son pequeños, por láser o electrocauterio, o si son grandes por cirugía.
Si el paciente, al momento de la exploración, presenta tumores de forma plexiforme, debe complementarse su estudio con resonancia magnética. Sin embargo, hay que tomar en cuenta que los resultados con tratamiento quirúrgico suelen presentar resultados insatisfactorios, debido a que los tumores, ya sea en su forma benigna o maligna, están relacionados con el sistema nervioso.
CONCLUSIÓN
El tratamiento de la neurofibromatosis tipo I se debe abordar desde un punto de vista integral, con el apoyo unido de los especialistas y los familiares, cuyo enfoque vaya encaminado a detectar signos y síntomas, a su abordaje y seguimiento clínico. Tomando en cuenta que en algunos casos se presentan patrones no tan alentadores (tumores malignos), el desenlace puede ser catastrófico para el paciente, familia y equipo de salud. Agregando a todo este cuadro de dolor y molestias consecuentes, un impacto emocional y físico a lo largo del proceso, donde el soporte por parte del especialista y de la medicina del dolor son fundamentales.
Por último, el trastorno de aprendizaje suele tener repercusiones importantes, sobre todo en pacientes pediátricos, los cuales se ven limitados en sus tres esferas, se debe indicar un refuerzo y acompañamiento de asesores en educación, ideales para un desarrollo integral en el paciente.
La neurofibromatosis tipo II es una enfermedad invalidante que se hereda de forma autosómica dominante. A menudo se ha confundido con la neurofibromatosis tipo I, aunque son patologías distintas.
*Universidad Nacional Autónoma de México, Tlalnepantla de Baz, México.
Contacto: fernandadeloya94@hotmail.com
**Universidad Anáhuac Mayab, Mérida, México.
Contacto: axelgb1@hotmail.com
REFERENCIAS
Bersusky, E. (2017). Deformidades vertebrales por neurofibromatosis tipo 1. Rev. Asoc. Arg. Ortop. y Traumatol. 64(4):263-269. Disponible en: https://www.aaot.org.ar/revis-ta/1993_2002/1999/1999_4/640401.pdf
Domínguez-Hernández, E. (2014). Neurofibromatosis tipo 1: enfermedad de von Recklin- ghausen. Sevilla, España: Inserm, p. Portal de información de enfermedades raras y medicamentos huérfanos.
Galán, E. (2014). Neurofibromatosis tipo 1. National Institute of Health, pp. 57-60. Disponible en: https://www.aeped.es/sites/default/files/documentos/10-nf1.pdf
García, R., Cervini, A., y Pierini, A. (2013). Manifestaciones cutáneas de la neurofibromatosis tipo 1. Arc. Argent. Pediatr. 101(2):127-132. Disponible en https://www.sap.org.ar/docs/publicaciones/archivosarg/2003/127.pdf
Gómez, M., y Batista, O. (2015). Neurofibromatosis tipo 1 y su diagnóstico molecular como estrategia del diagnóstico diferencial y a edades tempranas. Rev. Méd. Chile. 143(10):168-169. http://dx.doi.org/10.4067/S0034-98872015001000011
Jiménez-Caballero, P., López-Espuela, F., Portilla-Cuenca, J., et al. (2013). Manifestaciones clínicas y neurorradiológicas en los adultos con neurofibromatosis tipo 1. Neurología. 28(6):361-365. https://doi.org/10.1016/j.nrl.2012.09.001
Llorente-La Orden, C., Vidal-Villegas, B., Nárvaez-Palazón, C., et al. (2018). Manifestaciones oftalmológicas de neurofibromatosis tipo 1 en dos hermanas. Archivos de la Sociedad Canaria de Oftalmología. 29:112-113. Disponible en: https://dial- net.unirioja.es/servlet/articulo?codigo=6491251
Pardo-Somalo, R., y Málago-Guerrero, S. (2014). Hipertensión secundaria a coartación de aorta y estenosis de arteria renal en adolescente con neurofibromatosis tipo 1. Nefrología. 28(2):216-217. Disponible en: https://www.revistanefrologia.com/es-pdf-X021169950803303X
Ríos-Sanabria, C., y Mora-Hernández, G. (2014). Neurofibromatosis tipo 1: Enfermedad de von Recklinghausen. Revista Médica de Costa Rica y Centroamerica. LXXI (610):249-251. Disponible en: https://www.medigraphic.com/pdfs/revmedcoscen/rmc-2014/rmc142n.pdf
Salas-Alanís, J.C., De la Garza-Ramos, R., y Cepeda-Valdés, R. (2011). Neurofibromatosis tipo 1 (enfermedad de von Recklinghausen): reporte de dos casos. Dermatología CMQ. 9(4):268-271. Disponible en: https://www.medigraphic.com/pdfs/cosmetica/dcm-2011/dcm114e.pdf
Recibido: 23/08/2022
Aceptado: 02/02/2023