¿Qué es el síndrome de Parkes Weber?
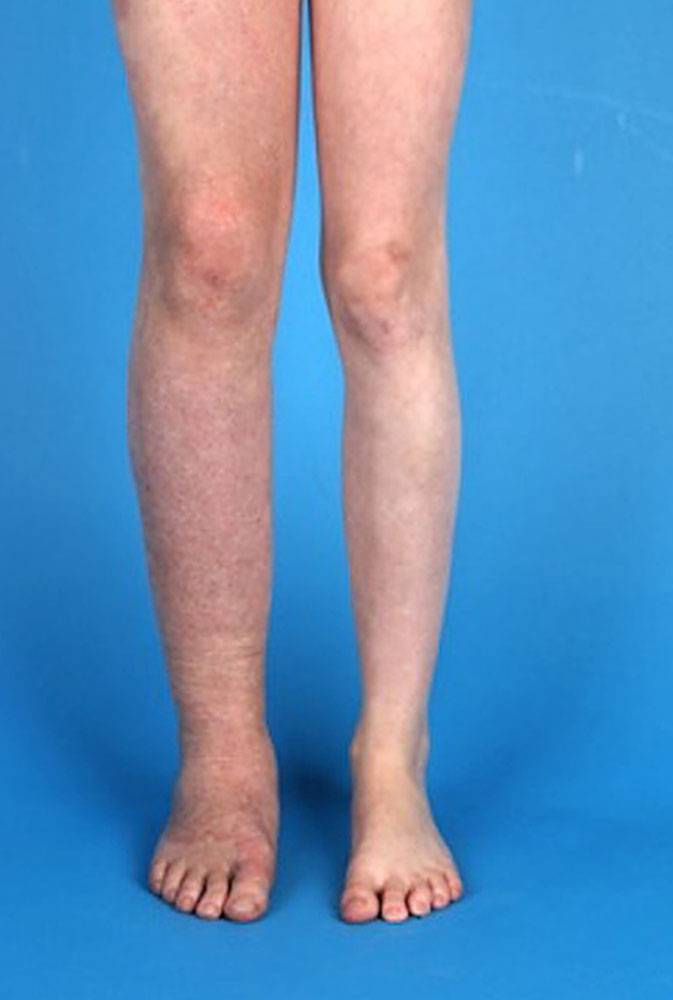
Se trata de una patología congénita, cuya incidencia es de 1 cada 100 mil personas, donde los afectados tienen malformaciones vasculares, varices, crecimiento excesivo de partes blancas y tejido óseo, además de manchas en vino de Oporto (coloraciones rojas, rosadas o violáceas presentes al nacer). En casos que muestran complicaciones se presenta insuficiencia cardiaca, celulitis, sangramientos y anemia.
Las causas son desconocidas y la condición es esporádica, es decir, no hay antecedentes del síndrome en familiares. Sin embargo, en ocasiones es causado por una mutación del gen RASA1 y se hereda de padres a hijos.
La enfermedad no tiene cura, y los tratamientos varían dependiendo de los síntomas que se presentan. Los problemas estéticos, como las manchas en la piel, pueden ser tratadas por un dermatólogo, mientras que las malformaciones y fisuras arteriovenosas requieren embolización o cirugía. Aunque hay lesiones en las que la cirugía no es posible y la embolización puede mejorar la calidad de vida durante un periodo de tiempo.
Gran parte de la morbimortalidad se debe a la cantidad de complicaciones y malformaciones de los órganos y sistemas. Los pacientes con obstrucción del drenaje linfático tienen mayor riesgo de fallecer por celulitis y bacteriemia.
Distintas organizaciones y grupos de apoyo proporcionan recursos valiosos a quienes padecen esta enfermedad, teniendo asesores médicos y clínicas expertas en el tema. Además de impulsar la investigación de nuevos tratamientos y posibles curas.
Ismael Medina
Referencias:
Avilez, C., y Zuniga, L. (2022). Síndrome de Parkes Weber en un neonato: reporte de un caso. Revista Hispanoamericana de Ciencias de la Salud. 8(3):99-104. Disponible en: file:///C:/Users/RevistaZJ/Downloads/Reporte_CasoF%20(1).pdf
Genetic and Rare Diseases Information Center. (2018). Síndrome de Parkes Weber. GARD, 15 de agosto. Disponible en: https://rarediseases.info.nih.gov/espanol/11991/sindrome-de-parkes-weber