MODELADO COMPUTACIONAL: UNA HERRAMIENTA PARA COMPRENDER LA CIENCIA DE LOS MATERIALES
CIENCIA UANL / AÑO 25, No.116, noviembre-diciembre 2022
Karla Silván-Díaz*, Guillermo Carbajal-Franco*
Para entender el impacto y las bondades del modelado de materiales mediante cálculos informáticos es necesario ubicar la trascendencia de los resultados obtenidos por estos métodos; la forma tradicional (hasta hace unos años) de investigación de materiales nuevos y sus propiedades era el trabajo en laboratorio con la inversión de recursos humanos y financieros. El modelado computacional cambia el paradigma introduciendo la experimentación mediante software, en el que se plantean sistemas y se prueban sus resultados a través de cálculos matemáticos, contribuyendo de esta forma a comprender la ciencia de los materiales. Una gran ventaja de esta forma de operación es que además de calcular la probabilidad de ocurrencia de una reacción, por ejemplo, proporciona datos de las propiedades de los materiales reactivos, así como de los producidos; información que tradicionalmente se obtiene de la caracterización llevada a cabo después de la obtención de éstos.
Para ilustrar lo anterior se utilizará como ejemplo la reacción de síntesis de la goetita FeO(OH), la fase alfa del hidróxido de hierro y principal precursora del óxido de hierro III, también en fase alfa, de gran valor por sus propiedades; estos últimos han cobrado relevancia como catalizadores y fotocatalizadores, cuyas reacciones presentan una gran compatibilidad con los métodos químicos empleados comúnmente para la elaboración de nanomateriales, además ofrecen una amplia gama de aplicaciones como catalizadores en procesos de oxidación química para tratamiento de aguas contaminadas (Shu-Sung y Mirat-Gurol, 1998), fotocatalizadores para fotodisociar la molécula de agua (Seriana, 2017; Long, et al., 2016), electrodepósitos para división de agua fotoelectroquímicamente (Arriaga-Arjona y Carbajal-Franco, 2017), entre muchas más.

El objetivo de la herramienta DMol³ basada en DFT (Density Functional Theory), incluida en el software Material Studio, es modelar procesos químicos y contribuir al entendimiento de las propiedades de los materiales de forma rápida y precisa, con un menor tiempo computacional, pero con una gran precisión de la Mecánica Cuántica. DMol³ está diseñada para brindar detalles de cálculos de potencia, de frecuencias vibratorias, propiedades termodinámicas, masa de los átomos y fuerzas de los enlaces, para identificar modos de curvatura negativa y localizar estados de transición.
El modelado molecular de procesos químicos es una alternativa para obtener detalles que no se logran a través de técnicas experimentales. La DFT es un método de cálculo de estructuras electrónicas, considera una nube electrónica (densidad de electrones), como se puede observar en la figura 1, en lugar de deducirlas por cada electrón y sus variables espaciales representadas en x, y, z. La DFT trae consigo el empleo de menor tiempo computacional al considerar la densidad electrónica en lugar de electrones individuales.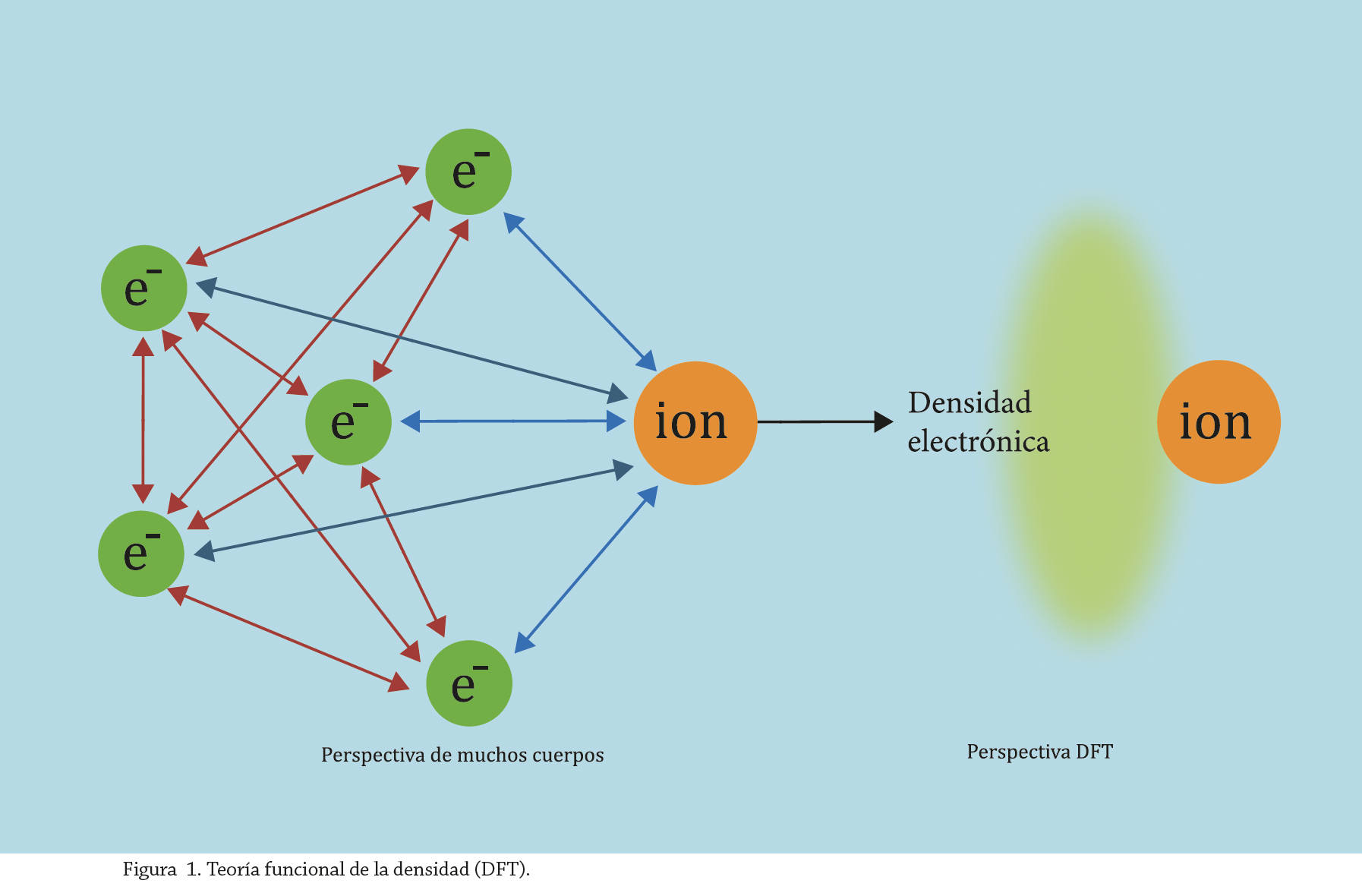
Una manera para determinar la interacción electrostática de la molécula con el disolvente es COSMO (Conductor-like Screening Model), un tipo de solvatación continua; en éste la molécula abre paso para crear una cavidad en el solvente, las distribuciones de carga polarizan dieléctricamente el medio y genera una polarización sobre la superficie de la cavidad. COSMO deriva las cargas de polarización a partir de una aproximación de conductor escalado, bajo la premisa de que si se conoce la distribución de carga eléctrica en la molécula, entonces es posible calcular la carga en segmentos de la superficie.
El estudio DOS (Density of States) es un concepto matemático y contribuye a entender los cambios causados por factores externos en estructuras electrónicas. Un cálculo de orbitales brinda información que incluye: orbital molecular ocupado más alto, HOMO (Highest Occupied Molecular Orbital), orbital molecular desocupado más bajo, LUMO (Lowest Unoccupied Molecular Orbital), y el spin del orbital. Durante el curso de una reacción química para obtener FeO(OH), la potencia total cambia naturalmente, comenzando con los reactivos va en aumento hasta un punto máximo y luego disminuye a la de los productos; en la activación será la máxima a lo largo de la reacción química para obtener FeO(OH); a la estructura correspondiente a ésta se le conoce como TS (Transition state); los estados de transición tienen altas energías, debido a que los enlaces deben romperse antes de formar nuevos (Maquez et al., 2016). TS es un punto estacionario, como un máximo de fuerza en dirección de la coordenada de reacción, en dicho punto se asume que los reactantes colisionan o se deforman a un estado inestable, dando paso a la formación de productos.
Para encontrar TS se aplican comúnmente algoritmos LST (Synchronous Transit Method) y QST (Quadratic Synchronous Method), que proporcionan estimaciones MEP (Mínimum Energy Pathway); una vez encontrado TS se puede realizar una optimización que dará oportunidad a predecir barreras de reacciones químicas y determinar vías de reacción (Solis Calero, 2013).
Investigaciones anteriores se han enfocado en identificar mediante DFT qué fase de FeO(OH) es más competente en la fotodisociación de agua (Zhenxiong et al., 2019); utilizando algoritmos LST/QST/CG calcularon una TS tardía endotérmica y de rápida cinética, y propusieron incorporar surfactantes para controlar el crecimiento de partículas (Maquez et al., 2016).
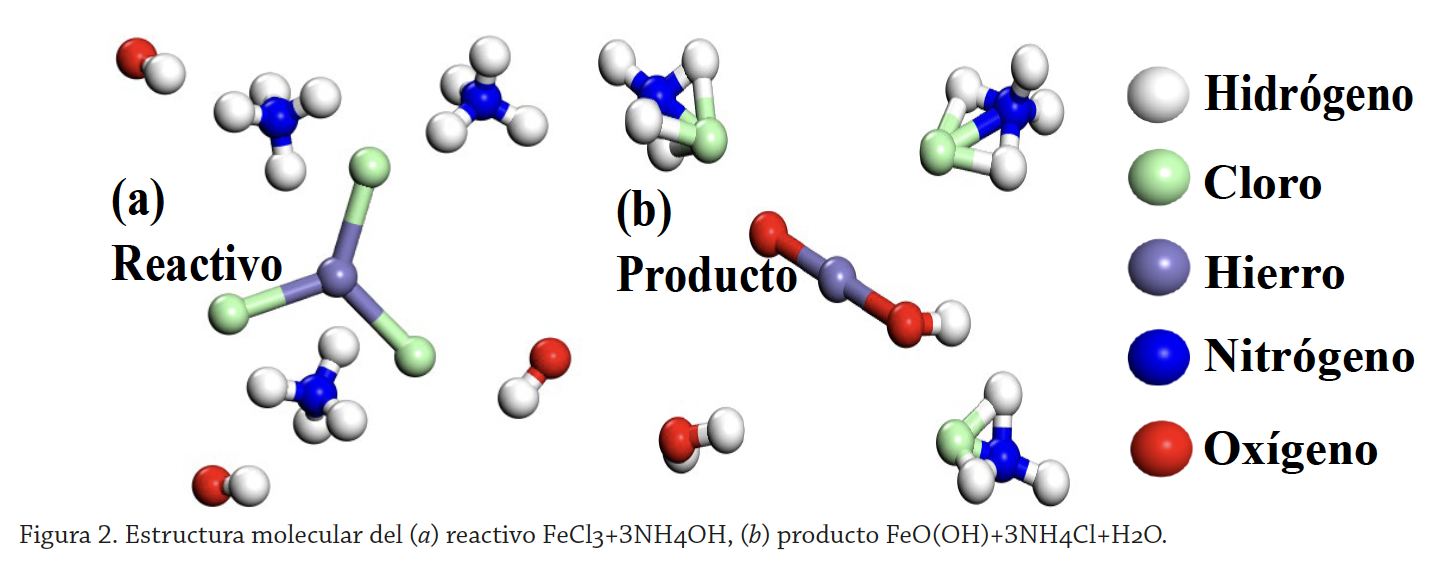
Es de suma importancia el análisis molecular de una reacción para obtener FeO(OH), ya que es uno de los óxidos de hierro más importantes empleados como adsorbentes y como soporte de catalizadores en incontables reacciones; en particular, la que se estudia en este trabajo se llevó a cabo mediante la reacción del cloruro de hierro III e hidróxido de amonio (figura 2a). El objetivo principal es obtener el estado de transición de una reacción para obtener FeO(OH) mediante DFT.
METODOLOGÍA
Todos los cálculos se realizaron con el paquete de programas DMol3 (McNellis et al., 2009). Se construyeron modelos del reactivo del producto (figura 2) optimizados geométricamente. La optimización geométrica fue llevada a cabo con una tolerancia de 1.0×10-4Ha, función de correlación de intercambio GGA-PBE, tolerancia SCF de 1.0×10-4eV, un orbital cutoff 3.5Å y utilizando COSMO, agua como solvente, con constante dieléctrica de 78.54. Para determinar la ruta de reacción TS se usaron los modelos de reactivo y producto optimizados, haciendo coincidir aquellos átomos equivalentes en de reactivo con el de producto. Para el cálculo TS se emplearon algoritmos LST/QST bajo las siguientes condiciones: protocolo de búsqueda completo LST/QST, convergencia RMS de 0.02 Ha/Å, función de correlación de intercambio GGA-PBE, tolerancia SCF de 1.0×10-4 eV, smearing de 0.015 Ha, agua como solvente con constante dieléctrica de 78.54.

RESULTADOS
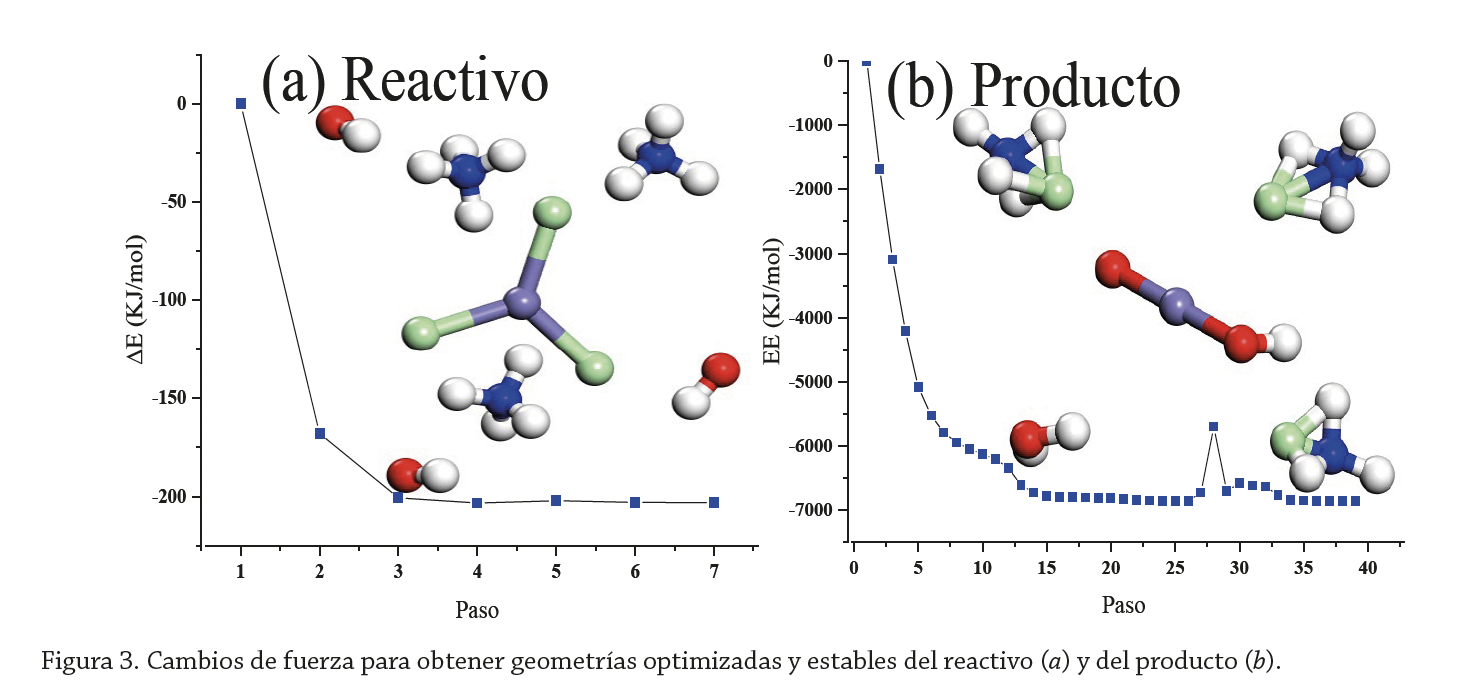
Las longitudes de los enlaces de las moléculas optimizadas son de 0.981 Å, 2.215 Å y 1.031 Å en los enlaces O-H, Fe-Cl, N-H, respectivamente, con ∆E=-0.07725 Ha (figura 3a) en los reactivos; mientras que las longitudes de los enlaces en el modelo del producto son de 1.676 Å, 1.823 Å, 0.981 Å, 1.033 Å y 1.305 Å para enlaces O-Fe, Fe-O, O-H, N-H y H-Cl, respectivamente, con ∆E=-2.60836 Ha (figura 3b). En comparación con lo reportado (Chatterjee, 2012), las longitudes de los enlaces O-H y N-H presentan una variación de ̴2%, el enlace H-Cl presenta una variación de ̴3%, y el enlace Fe-Cl presenta una variación de ̴35%.
El orbital molecular HOMO tiene una energía de -0.131 Ha o -3.554 eV; el LUMO, 0.0112 Ha o 0.303 eV. La diferencia entre la estructura molecular del reactivo y la estructura molecular del estado de transición es ∆E=0.03689 Ha (energía de activación), con una coordenada de reacción 0.48, y la diferencia entre el reactivo y el producto es ∆H= -0.08970 Ha (figura 4). Crayton (2002) reporta el comportamiento molecular mediante estados de transición de la goetita con una energía de activación con velocidad asociada al TS de 2̴ 8±11KJ/mol. Navakova et al. (2012) obtuvieron goetita mediante precipitación química del FCl3 con adición de sustancias tensoactivas; para explicar que el tamaño y crecimiento de las partículas de goetita afectan la adición de sustancias, llevaron a cabo estudios de espectroscopía y análisis termomagnético.
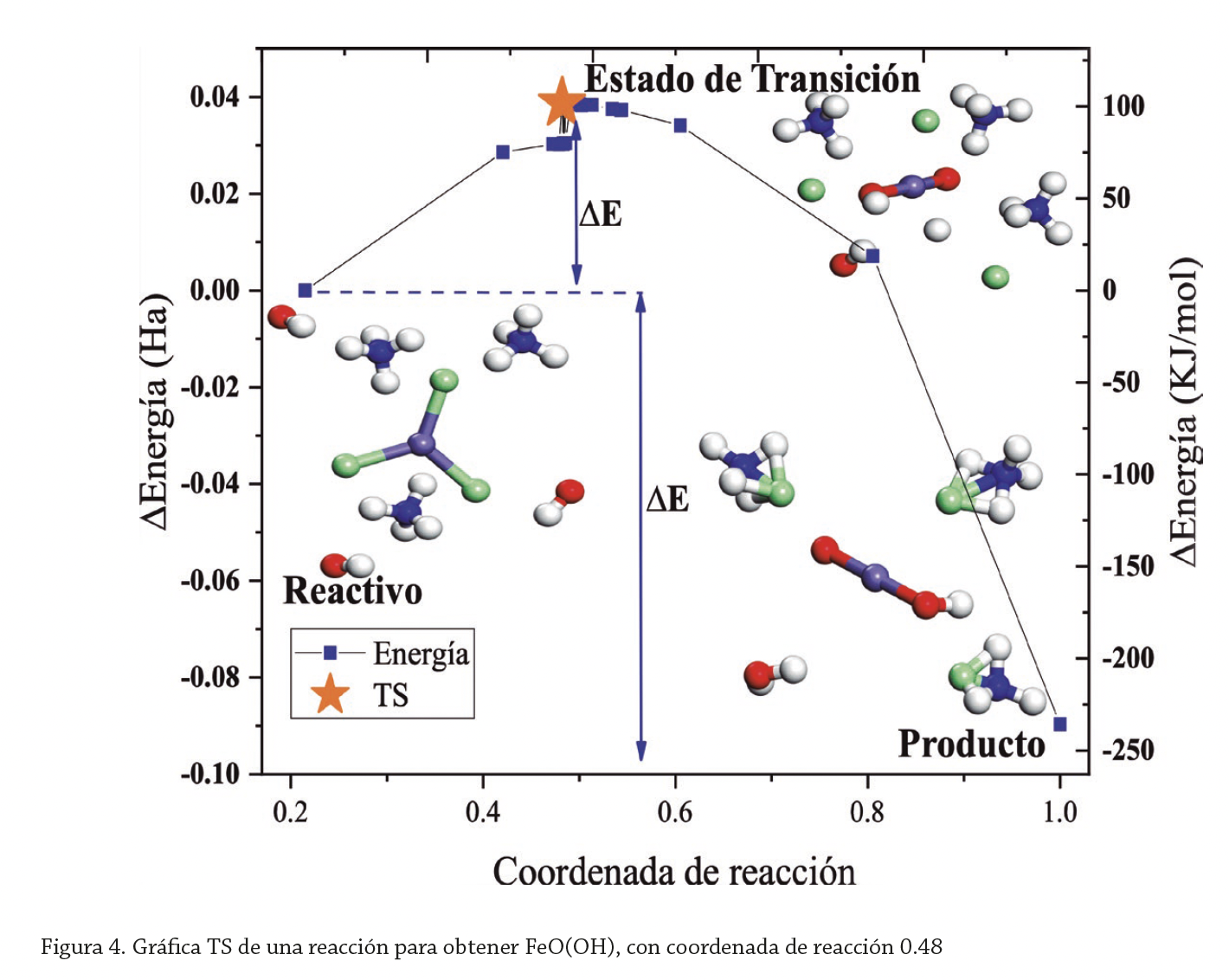
El TS de nuestro sistema presentó un nivel más alto de energía que los reactivos y los productos; la diferencia o el incremento de ésta entre el reactivo y el TS (∆E) es relativamente muy pequeña; las moléculas reaccionantes son energéticamente favorecidas para escalar la barrera de potencia (0.03689 Ha), y llegar de una forma rápida al TS.

CONCLUSIONES
Mediante el uso del programa DMol³ se realizó el modelado molecular de una reacción para obtener FeO(OH); los resultados concuerdan grandemente con los experimentales de laboratorio.
Se atribuyen los resultados al uso del método COSMO, se empleó agua como solvente. Tomando en consideración que la diferencia entre la energía potencial del reactivo y la del producto es negativa, se concluye que es una reacción endotérmica, el sistema simulado absorbió calor del entorno. Los incrementos de la misma que mostró el sistema de reactivos y productos nos proporciona información adicional, la cual considera que es fácil regresar de un estado final a uno inicial (reacción reversible), ya que, como era de esperarse, el TS de nuestro sistema está en un nivel más alto de fuerza que la de los reactivos y la de los productos; se considera este incremento entre el reactivo y el TS relativamente muy pequeño, se concluye que las moléculas son favorecidas para escalar la barrera energética y llegar fácilmente a un estado de transición.
Comparando los resultados reportados en Maquez et al. (2016), quienes reportan un mayor uso de energía de transición; el menor uso de ésta se atribuye a que se empleó como solvente implícito en COSMO, las distribuciones de cargas del soluto polarizan el medio dieléctrico de la superficie, por lo que disminuye la demanda para la obtención de FeO(OH).
La realización de este trabajo pone de manifiesto la importancia de la aplicación del modelado molecular como una herramienta de aprendizaje a bajo costo para entender la química a niveles básicos. La DFT es una teoría que coloca en la cima los avances en las soluciones aproximadas de la rama de la ciencia que describe el comportamiento de la materia a nivel microscópico (Mecánica Cuántica).

Al Dr. Guillermo Carbajal Franco por brindarnos todo su conocimiento y ofrecernos el equipo necesario para desarrollar el modelado computacional aplicado; al Conacyt por la beca otorgada, al TecNM y al ITTOL por el apoyo brindado y a la familia, por ese apoyo incondicional.
* Tecnológico Nacional de México/Instituto Tecnológico de Toluca, Metepec, México. Contacto: ksilvand1@toluca.tecnm.mx
REFERENCIAS
Arriaga-Arjona, L., y Carbajal-Franco, G. (2017). Zinc oxideiron-aluminum nanostructured cover for photoelectrchemical water splitting. Materials Research Society. 2:2707-2711.
Chatterjee, S. (2012). Encyclopedia of Inorganic Chemistry. West Bengal: Discovery Publishing House PVT. LTD.
Crayton, J. (2002). A model for 18O/16O variations in CO2 evolved from goethite during the solid-state FeOOH to Fe2O3 phase transition. Geochimica et Cosmochimica. 67(11):1991-2004.
Katrin, O., Wolfgang, W., y Schmahí, R. (2012). Density functional theory study of water adsorption on FeOOH surfaces. Germany: Surface Science.
Long, Z., Fangyuan, X., y Meng, Q. (2016). The adsorption and dissociation of water molecule on goethite (010)surface: A DFT approach. Applied Surface Science. 392:760-767.
Maquez, M., Carbajal-Franco, G., y Pacheco, J. (2016). Estado de transición de la reacción de sintesis de goetita mediante simulacion molecular. Revista de Simulación y Laboratorio. 3:24-32.
McNellis, E., Meyer, J., y Baghi, A.D. (2009). Stabilizing a molecular switch at solid surfaces: A density functional theory study of azobenzene on Cu(111), Ag(111), and Au(111). Physical Review B. 80(3):3-15.
Novakova, A., Antonov, A., Gendler, T., et al. (2012). The influence of surface active substances various concentrations on goethite nanoparticles magnetic properties. Solis State Phenomena. 190:447-450.
Seriana, N. (2017). Ab Initio simulations of water spliting on hematite. Journal of Physics: Condensed Matter. 29(46):3002.
Shu Sung, L. y Mirat Gurol , D. (1998). Catalytic Decomposition of Hydrogen Peroxide on Iron Oxide: Kinetics, Mechanism, and Implications. Environmental Science & Technology. 32(10):1417-1423.
Zhenxiong, H., Fengshuang, H., Mingtao, L., et al. (2019). Which phase of iron oxyhydroxides (FeOOH) is more competent in overall water splitting as a photocatalyst, goethite, akaganeite or lepidocrocite? A DFT-based investigation. China: Computational Materials Science.