Mapas de potencial electrostático para la comprensión de la reactividad química
SARAÍ VEGA RODRÍGUEZ*
CIENCIA UANL / AÑO 25, No.113, mayo-junio 2022
El estudio de las reacciones químicas es en gran medida el corazón de la Química. Una reacción química consiste en una transformación en la que inicialmente se tiene un conjunto de especies químicas (átomos, moléculas, compuestos químicos, etc.) que interactúan entre ellas para finalmente formar especies diferentes a las iniciales (Kotz et al., 2014). La facilidad o capacidad para reaccionar se conoce como reactividad. La reactividad depende de varios factores, siendo la naturaleza de la especie química el factor más relevante. Si conocemos o comprendemos la naturaleza de las especies químicas podemos entender o predecir de qué manera reaccionarán; sin embargo, para algunos compuestos no es tan sencillo. Una manera de ayudar a visualizar la reactividad de una molécula es a través de los mapas de potencial electrostático.
Un mapa de potencial electrostático (MPE) muestra la fuerza de atracción o repulsión que experimenta una partícula, cargada positivamente, cuando interactúa con la superficie de una molécula (Hardinger, 2010). El MPE nos indica, mediante una escala de colores, cuáles son las regiones de la molécula que carecen o que tienen un exceso de densidad electrónica. Podemos entender la densidad electrónica como una nube de electrones en la molécula. Esta información es de gran importancia para los químicos, porque a partir de ella se puede conocer la reactividad de una molécula, y esto permite diseñar metodologías para la síntesis de compuestos orgánicos e inorgánicos.
¿CÓMO SE RELACIONA EL MPE CON LA REACTIVIDAD DE UNA MOLÉCULA?
Las regiones que carecen de densidad electrónica son susceptibles al ataque de especies que contienen electrones; las especies que tienen un exceso de densidad electrónica atacan a las especies que carecen de ella. Estos “ataques” forman enlaces que llevan a la generación de compuestos químicos.
En Química Orgánica se denomina nucleófilo a los reactivos que forman enlace al donar sus electrones a especies carentes de densidad electrónica o electrófilo (McNaught y Wilkinson, 2019). Entonces, por medio de un MPE es posible saber si una molécula, o una región de esta, se comporta como electrófilo o como nucleófilo. Esto, finalmente, permite predecir cómo reaccionará esa molécula con otras.
Los MPE también permiten visualizar el efecto que tienen diferentes grupos funcionales en una molécula. Si tomamos como ejemplo un anillo de benceno y cambiamos un átomo de hidrógeno por otro sustituyente (grupo funcional), podremos observar cómo se modifica la densidad electrónica del benceno. En la figura 1 se presentan los MPE de benceno ![]() metilbenceno
metilbenceno ![]() aminobenceno
aminobenceno ![]() cianobenceno
cianobenceno ![]() y nitrobenceno
y nitrobenceno ![]() En la escala de colores, el rojo indica zonas de alta densidad electrónica que se va desvaneciendo al pasar por naranja, amarillo y verde, hasta llegar al azul que indica zonas carentes de densidad electrónica.
En la escala de colores, el rojo indica zonas de alta densidad electrónica que se va desvaneciendo al pasar por naranja, amarillo y verde, hasta llegar al azul que indica zonas carentes de densidad electrónica.
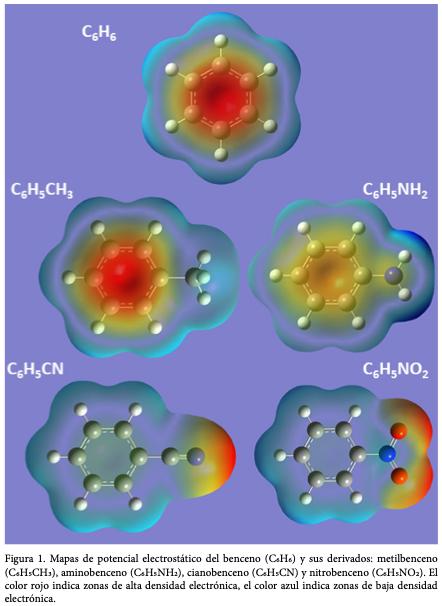
De acuerdo con el MPE de benceno ![]() la densidad electrónica se agrupa en el centro del benceno porque ahí se encuentran los electrones pi que forman el anillo aromático.
la densidad electrónica se agrupa en el centro del benceno porque ahí se encuentran los electrones pi que forman el anillo aromático.
La presencia de los grupos nitro y ciano reduce la densidad electrónica en el anillo aromático, esto se puede observar en el mapa de las moléculas de cianobenceno ![]() y nitrobenceno
y nitrobenceno ![]() , en las que la densidad electrónica ahora se localiza en el grupo funcional. Esto se debe a que estos grupos son electroatractores (jalan densidad electrónica por medio de resonancia o efecto inductivo) y dejan el anillo susceptible a un ataque nucleofílico.
, en las que la densidad electrónica ahora se localiza en el grupo funcional. Esto se debe a que estos grupos son electroatractores (jalan densidad electrónica por medio de resonancia o efecto inductivo) y dejan el anillo susceptible a un ataque nucleofílico.
APLICACIÓN AL ENTENDIMIENTO DE UNA REACCIÓN QUÍMICA: SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA (SEAr)
Una reacción de sustitución electrofílica aromática (SEAr) es aquélla en la que los electrones del anillo aromático atacan a un electrófilo, de manera que esta especie se une a un átomo de carbono del anillo aromático, mientras que el hidrógeno unido a ese carbono sale, de manera que el electrófilo sustituye o reemplaza a un átomo de hidrógeno del anillo aromático (figura 2). De acuerdo con el MPE, el benceno concentra la densidad electrónica en el anillo aromático; por lo tanto, es un nucleófilo o una especie rica en electrones que puede reaccionar con electrófilos o especies carentes de electrones. Cuando el benceno tiene sustituyentes diferentes a los átomos de hidrógeno, la SEAr se ve afectada.
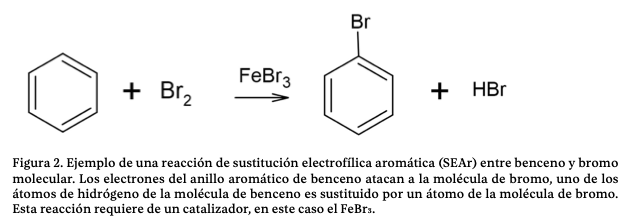
Los sustituyentes electroatractores (ciano, nitro) retiran o disminuyen casi por completo la densidad en el anillo, desactivándolo a la SEAr. Se sabe que el nitrobenceno es 100,000 veces menos reactivo que el benceno en lo que se refiere a la SEAr (Wade, 1993). En cambio, los sustituyentes electrodonadores (amina, metilo) aportan a la densidad electrónica del anillo, activándolo para la SEAr. El metilbenceno es 25 veces más rápido que el benceno para la SEAr (Wade, 1993).
OBTENCIÓN DE LOS MPE
Para obtener el MPE de una molécula se hace uso de herramientas de Química computacional a partir de las cuales se puede modelar una molécula y calcular sus propiedades. Específicamente, se utilizan programas especializados que se basan en la resolución aproximada de la ecuación de Schrödinger, de manera que nos permiten conocer la estructura electrónica de una molécula y, por ende, sus propiedades. En el cálculo de los MPE se modela una partícula con carga positiva que recorre la superficie de una molécula. Esta partícula puede experimentar atracción hacia las regiones cargadas negativamente, o repulsión hacia las regiones cargadas positivamente.
La energía de atracción o repulsión de la molécula con esa carga se expresa mediante la ecuación (1), donde V(r) es el potencial electrostático generado por la interacción de la molécula con una carga situada a una distancia r; Z es el número atómico de cada átomo A en la molécula; (r’) es la densidad electrónica de la molécula; RA-r es la distancia entre la carga y los núcleos y r´-r es la distancia entre la densidad electrónica de la molécula y la carga (Politzer et al., 2009).
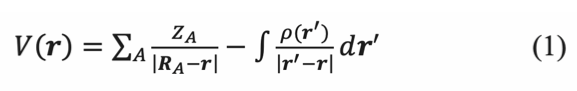
Los MPE mostrados en este artículo fueron calculados al nivel de teoría HF/6-31G, es decir, con el método ab initio Hartree-Fock (Roothaan, 1951) y el conjunto de bases 6-31G (Binkley et al., 1980), utilizando el paquete de programas de estructura electrónica Gaussian09W (Frisch et al., 2010).
CONCLUSIONES
Los MPE son útiles para la comprensión de la reactividad de una molécula, éstos pueden ser utilizados como un complemento para entender los conceptos de electrofilicidad y nucleofilicidad, incluso son utilizados como parte de la metodología de diseño de fármacos. Cabe mencionar que los MPE son una primera aproximación para visualizar la reactividad, ya que existen metodologías más adecuadas para estudiar la reactividad de una molécula, como el cálculo de índices de reactividad globales: potencial químico, electronegatividad, blandura, dureza; o locales: densidad electrónica y funciones de Fukui (Pearson, 2005). La ventaja de los MPE sobre estas metodologías reside en la simplicidad para obtenerlos.
* Universidad Autónoma de San Luis Potosí, San Luis Potosí, México.
Contacto: sarai.vega@uaslp.mx
REFERENCIAS
Binkley, J.S., Pople, J.A., y Hehre, W.J. (1980). Self-Consistent Molecular Orbital Methods. 21. Small Split-Valence Basis Sets for First-Row Elements. J. Am. Chem. Soc. 102:939-47.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. (2010). Gaussian 09, Revision C.01. Gaussian, Inc., Wallingford CT.
Hardinger, S.A. (2010). Illustrated Glossary of Organic Chemistry. Disponible en: http://www.chem.ucla.edu/~harding/IGOC/E/electrostatic_potential_map.html#:~:text=Electrostatic%20potential%20map%3A%20A%20map,electron%20excess%20and%20electron%20deficiency
McNaught, A.D., y Wilkinson, A. (2019). Compendium of Chemical Terminolog (the «Gold Book»). IUPAC Recomendations. Blackwell Scientific Publications, Oxford. Disponible en https://doi.org/10.1351/goldbook
Kotz, J.C., Treichel, P.M., Townsend, J., et al. (2014). Chemistry & chemical reactivity. Cengage Learning.
Pearson, R.G., (2005). Chemical hardness and density functional theory. J. Chem. Sci. 117(5):369-377.
Politzer, P., y Murray, J.S. (2009). The Electrostatic Potential as a Guide to Molecular Interactive Behavior Chemical Reactivity Theory-A Density Functional View, CRC Press.
Roothaan, C.C.J. (1951). New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 23:69.
Wade L.G. (1993). Química Orgánica. México: Pearson Education.