Síndrome de Sturge Weber/angiomatosis encefalotrigeminal. Una revisión
Axel García-Burgos*, Michelle Danairy Canudas-Zertuche**,
José Eduardo Campos-Arceo***
CIENCIA UANL / AÑO 24, No.108, julio-agosto 2021
RESUMEN
El Síndrome de Sturge Weber forma parte de las enfermedades clasificadas como facomatosis. Es un síndrome poco frecuente que engloba distintos sistemas del cuerpo humano, como el tegumentario. Se define como “un síndrome que engloba anomalías cerebrales, cutáneas y oculares” (Fernández-Jaén, Sánchez-Jacob y Ramos-Boludac, 2004). Se presenta de manera esporádica, afecta la “microvasculatura venosa cefálica, agregando la manifestación cutánea, la cual se presenta como angioma facial plano de color rojo vino conocida también como mancha de vino Oporto” (Fernández- Concepción, Gómez-García y Hernández-Sardiñaz, 1999). Ésta puede presentar glaucoma y alteraciones oculares. De la misma manera, dado su afectación en el sistema nervioso, pueden resentarse episodios epilépticos, hemiparesia facial, retraso en el desarrollo de aprendizaje y trastorno por déficit de atención e hiperactividad (Morales-Querol, Sierra-Benítez y Márquez-Oquendo, 2017). por lo que este padecimiento debe englobar una intervención multidisciplinaria en la que debió realizar una revisión de cada manifestación para llegar a un tratamiento beneficioso para el paciente.
Palabras clave: facomatosis, tegumentario, angioma, glaucoma, hiperactividad.
ABSTRACT
Sturge Weber syndrome is one of the diseases classified as phakomatosis. It is a rare syndrome which involves different systems of the human body, including the integumentary system. Sturge Weber syndrome is defined as: “a syndrome that encompasses brain, skin and eye abnormalities” (Fernández-Jaén, Sánchez-Jacob y Ramos-Boludac, 2004). It occurs sporadically, affects the cephalic venous microvasculature, the cutaneous manifestation appears as a wine-red flat facial angioma (Oporto Wine Stain) (Fernández-Concepción, Gómez-García y Hernández-Sardiñaz, 1999), can appear glaucoma and eye alterations, epileptic episodes, facial hemiparesis, delayed learning development, and hyperactivity (Morales-Querol, Sierra-Benítez y Marquez-Oquendo, 2017) disorder may occur. Therefore, this condition must have a multidisciplinary intervention in which each manifestation must be analyzed to reach a convenient treatment for the patient.
Keywords: phakomatosis, integumentary, angioma, hiperactivity.
El Síndrome de Sturge Weber, antes nombrado como angiomatosis encefalotrigeminal por “WilliamAllen Sturge en 1879 y Frederick Parker Weber en 1922, tiene manifestaciones clínicas: anomalías cerebrales (angioma leptomeníneo o pial), cutáneas (angioma facial) y oculares (angioma coroideo)” (Morales-Querol, Sierra-Benítez y Marquez-Oquendo, 2017). En relación con este síndrome se han descrito tres variaciones, “tipo I: angiomatosis leptomeníngea, en ésta el paciente puede presentar glaucoma. Tipo II: se presenta angioma facial, el sistema nervioso central no se encuentra afectado, puede presentarse glaucoma. Tipo III: se presenta angiomatosis leptomeníngea, sin presentarse glaucoma” (Cerisola-Cardoso et al., 2008). En las fases tempranas de la edad es conveniente realizar una revisión y exploración por parte del especialista en pediatría y neurología en estas fases tempranas de la enfermedad. Cabe mencionar que los hallazgos clínicos deben clasificarse de acuerdo a los criterios anteriormente mencionados.
ETIOLOGÍA DEL SÍNDROME DE STURGE WEBER
Es importante hacer hincapié en que la etiología radica en una mutación somática, en el “gen GNAQ, localizado en el cromosoma 9, en la región 21, ésta a su vez codifica para la proteína Gq alfa, la cual se encarga de realizar el proceso de señalización celular, para la comunicación de neurotransmisores y responsable de factores de crecimiento, vasoactivos y neurológicos” (Núñez-Gamboa, 2018), esto explica la falla y su repercusión en los sistemas cutáneos, nerviosos y oculares. Además influye en el factor del crecimiento, tanto en el aprendizaje y en el trastorno por déficit de atención por hiperactividad. Según informes recientes, se deben realizar los estudios correspondientes, como se explicará más adelante, durante el periodo prenatal y desarrollo preescolar para valorar la evolución y la historia natural de la enfermedad en el paciente.
Epidemiología
Su incidencia se estima en: “1 cada de 50,000 recién nacidos vivos, afecta de manera igual en ambos sexos. A nivel mundial, no tiene predominancia especial por algún grupo étnico” (McBride, 2017). Por lo anteriormente descrito, se diagnostica en los primeros meses de vida del paciente. Se ha registrado una variante de la angiomatosis leptomeníngea sin la presencia del nervio facial.
Embriología
Diversos estudios y observaciones en los pacientes pediátricos han permitido entender el origen de la enfermedad de la angiomatosis leptomeníngea. “Se debe a un fallo en la regresión del plexo venoso cefálico primitivo” (Higueros et al., 2017). Anatómicamente, durante el proceso de formación del ser humano, el sistema venoso primitivo se divide en “porción externa, que abastece y drena la piel de la cara y del cuero cabelludo; una porción media, que irriga las meninges, y una porción profunda, que abastece y drena el cerebro” (Higueros et al., 2017). Por lo cual el sistema de irrigación sanguíneo se altera ocasionando la aparición de episodios neurológicos como cefalea intensa, dificultad y desarrollo del lenguaje y trastornos del aprendizaje.Asimismo, de acuerdo a la situación anatómica, la formación del ectodermo será la base del “área superior de la piel de la cara con la parte del tubo neural que formará el área parieto-occipital del cerebro” (Higueros et al., 2017), esto podría explicar la asociación y aparición cutánea de la mancha de vino Oporto facial con el angioma leptomeníngeo, conformando el Síndrome de Sturge Weber.
MÉTODOS DE DIAGNÓSTICO (MATERIALES Y MÉTODOS)
El diagnóstico principal se basa en los hallazgos clínicos del paciente, por lo que dependiendo de la “extensión de marca de nacimiento (mancha de vino Oporto), el riesgo puede encontrarse entre 15-45% de padecer el Síndrome de Sturge Weber” (Samra y Portillo, 2004). Por otro lado, el diagnóstico se complementa con estudios de imagen (radiografía, tomografía computarizada o resonancia magnética con contraste gadolinio). Mediante el progreso de la enfermedad se puede observar: “hemiatrofia cerebral ipsilateral, calcificaciones corticales y delinean circunvalaciones cerebrales y angiomatosis leptomeníngea” (Morales-Querol, Sierra-Benítez y Marquez-Oquendo, 2017). Por otro lado, también las imágenes cerebrales pueden mostrar alteraciones en el flujo sanguíneo cerebral.
Signos y síntomas del Síndrome de Sturge Weber
En numerosos artículos y guías consultadas se tiene documentada una variedad de manifestaciones clínicas como “la aparición de mancha de coloración de vino de Oporto, tiene una presentación generalmente en el nacimiento, localizado en la frente, en el párpado superior en uno o ambos lados de la cara” (Martínez-Gutiérrez, López-Lancho y Pérez-Blázquez, 2008). Como se mencionó en el apartado anterior, afecta también en el desarrollo y crecimiento del paciente afectado, debido a esto puede presentar: “hipertrofia de tejido blando y óseo, los cuales pueden llevar a problemas de la audición, visión, deglución y habla” (Samra y Portillo, 2004). La afectación de la visión se presenta en 50% de los pacientes, quienes pueden desarrollar glaucoma, y lleva a atrofia óptima y ceguera de manera progresiva. Los lactantes pueden “presentar episodios de epilepsia, desviación de la mirada, esto ocasionado por la angiomatosis leptomeníngea, debido a que el nervio trigémino es afectado” (Ríos, Barbot y Pinto, 2012).
Manifestaciones cutáneas
Las lesiones cutáneas de la enfermedad presentan un patrón: “angioma plano congénito de la cara (mancha de vino Oporto), generalmente no progresivo, ni recesivo, localizado a nivel del parpado superior y frente” (Morales-Querol, Sierra-Benítez y Marquez-Oquendo, 2017). Suele presentarse de un mismo lado, puede extenderse a otras zonas de la piel y mucosas. Dichas alteraciones cutáneas pueden encontrarse al momento de realizar la exploración física en el paciente pediátrico desde el nacimiento hasta la etapa aproximada de los 12 años, por lo cual necesitará monitoreo continuo para observar los patrones de crecimiento de la enfermedad.
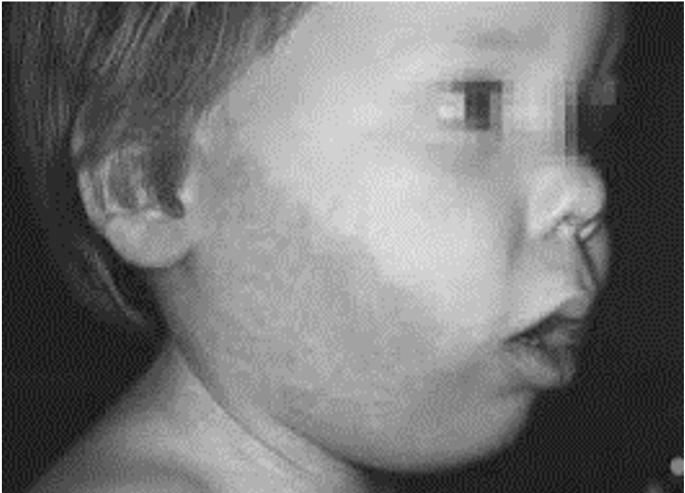
Figura 1. El patrón característico de esta enfermedad son las manchas de vino Oporto que aparecen en la región facial; pueden aparecer a nivel del párpado superior y en otras zonas de la piel, principalmente en pacientes pediátricos (antes de los cinco años) (Iderma.es, 2016)
Manifestaciones neurológicas
Dentro de la aparición y desarrollo de los signos y síntomas neurológicos, se puede observar “hemiparesia lentamente progresiva, se presenta de 25 a 60% de los pacientes, ocasionado por la atrofia cerebral, retraso del desarrollo psicológico y de aprendizaje en 45 a 60% de los pacientes, de la misma manera esto ocasiona alteraciones en la conducta y desarrollo social del paciente pediátrico. Pueden presentarse cefaleas de tipo migrañoso en un 30%” (Martínez-Gutiérrez, López-Lancho y Pérez-Blázquez, 2008). Se recomienda a los padres y jefes de familia realizar una revisión detallada en los pacientes y seguimiento por parte de especialistas en psicología y educación, debido a que el desarrollo de los pacientes suele ser difícil en su entorno y por tanto el entorno familiar y escolar representa un reto para el niño o joven. A su vez, requiere también valoración por parte de neurólogo y de imagenología, la valoración del área cerebral también puede arrojar “calcificaciones corticales cerebrales (Akhter-Kainat et al., 2014).
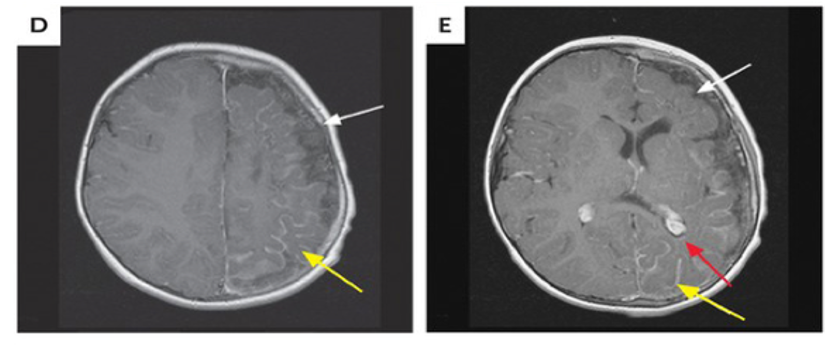
Figura 2. Se observan las calcificaciones y alteraciones en el flujo sanguíneo, éstas causan alteraciones neurológicas y del estado del paciente. Síntomas como la cefalea tipo migrañosa y alteraciones en el aprendizaje son causadas por las calcificaciones (NJEM, 2017).
Manifestaciones oftálmicas
Existen alteraciones a nivel oftálmico, sumado al deterioro neurológico, éstas se presentan de manera progresiva. La afectación visual comienza en el paciente pediátrico y la sintomatología continúa en el paciente joven. Por casos clínicos y bibliografía, se ha documentado que “más de 50% de los pacientes pediátricos desarrolla atrofia óptica, glaucoma y ceguera” (Akhter-Kainat et al., 2014). Además, se ha observado que se desarrolla de manera simultánea “angioma coroideo-retiniano a nivel de la esclera” (Hernández y Herrera, 2019). Por esto es importante la detección temprana y seguimiento, lo que permite tomar medidas, como se verá más adelante, en torno al paciente.
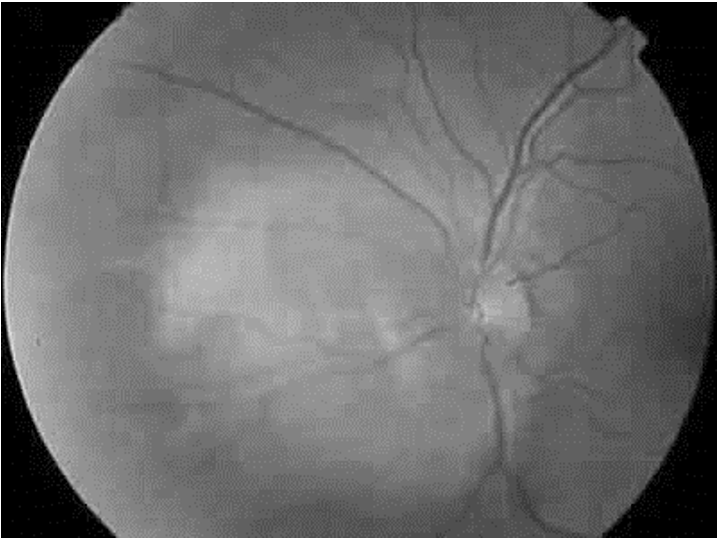
Figura 3. Angioma circunscrito tratado con terapia fotodinámica en un paciente de 14 años (Scielo España, 2008).
Manifestaciones faciales
Durante la exploración física en el paciente se pueden observar ciertas características faciales: “hendidura situada en la frente, mala oclusión y clinodactilia del quinto dedo en una de las extremidades superiores” (Steve-Roach, 2020). Estos signos clínicos ya analizados e inspeccionados por el profesional de salud facilitan la atención y la definición del caso a tratar.
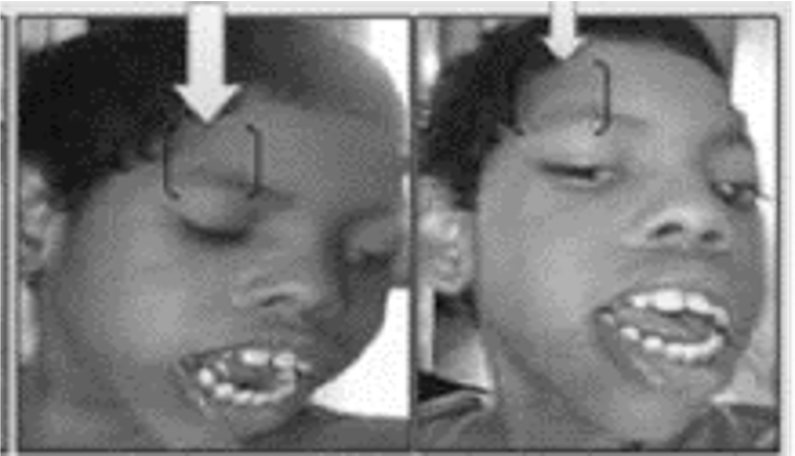
Figura 4. Al momento de realizar la exploración física, pueden notarse ciertas expresiones o características faciales, las cuales pueden estar vinculadas con el Síndrome de Sturge Weber, al igual que alteraciones odontológicas como una malaoclusión en el paciente pediátrico y juvenil (NEJM, 2017).
Diagnóstico diferencial en el Síndrome de Sturge Weber
El diagnóstico diferencial se basa en la revisión de los signos y síntomas que se presenten en la evolución de la enfermedad. Como se mencionó anteriormente, existen tres variaciones (clasificación de Roach): tipo I, tipo II y tipo III.
Dentro de la clasificación de tipo I “se presentan lesiones cutáneas como el angioma facial (mancha de vino de Oporto) y leptomeninge. Existe posibilidad de que el paciente pueda presentar glaucoma. Dentro de la clasificación de tipo II se presenta la lesión facial (mancha de vino de Oporto). El sistema nervioso del paciente no se encuentra comprometido. Existe posibilidad que el paciente pueda presentar glaucoma. Dentro de la clasificación de tipo III el paciente presenta el sistema nervioso comprometido. No existe riesgo de glaucoma y afectación al sistema ocular” (Maraña-Pérez et al., 2016).
Tratamiento en el Síndrome de Sturge Weber
Para el tratamiento principal de la angiomatosis se requiere una revisión exhaustiva del sistema nervioso central, oftalmológico y dermatológico. Los pacientes presentan crisis convulsivas, episodios de déficits focales, glaucoma, cefalea y trastorno del desarrollo. En primer lugar, la existencia de un déficit focal transitorio en ausencia de crisis convulsiva. En segundo lugar, hallazgos radiológicos, como las calcifi- caciones. Por lo que el tratamiento de elección es “el uso de antigregante con ácido acetilsalicílico. Se emplean anticonvulsivos y fármacos para tratar las convulsiones y glaucoma. Se han documentado ciertos efectos secundarios de algunos fármacos como el topiramato, por su efecto secundario se ha observado la aparición de glaucoma agudo bilateral, pudiendo empeorar el diagnóstico en el paciente” (Maraña-Pérez et al., 2016).
Se han encontrado distintos resultados por medio de la terapia quirúrgica para los pacientes que presentan glaucoma. “La trabeculectomía ha sido empleada en 61.5%, frente a la terapia fotodinámica empleada en 38.5% de los pacientes” (Rodríguez-Osorio et al., 2018).
En ocasiones puede realizarse la hemisferectomía debido a que pueden presentarse crisis convulsivas intratables, representando una mejoría importante por parte de los pacientes y en sus crisis convulsivas. En ésta se ha “reportado una desaparición de las crisis, desde la realización de la cirugía hasta los 7 meses del seguimiento de los resultados” (Rodríguez-Osorio et al., 2018). Específicamente en el angioma facial (mancha de vino Oporto), puede realizarse “fototermólisis, para disminuir la lesión cutánea de color violáceo, con resultados favorables para el paciente, por lo cual puede tener un mejor desarrollo en su medio”. Asimismo, se recomienda a los padres del paciente evitar la exposición fotodinámica a los factores de la luz UV y solar. Conviene realizar un seguimiento psicológico y educacional en el paciente, debido a que puede presentar severos trastornos del lenguaje, desarrollo y aprendizaje en su medio social.
DISCUSIÓN
El abordaje del Síndrome de Sturge Weber debe realizarse desde un punto de vista integral. Debe existir un trabajo interdisciplinario por parte de los especialistas en dermatología, oftalmología, neurología y psicología/desarrollo-aprendizaje, basándose en los signos y síntomas que presente el paciente. Un factor de suma importancia es la edad de aparición de las manifestaciones clínicas, ya que el paciente comienza a experimentarlo desde sus primeros meses de vida.
Tomando en cuenta que el abordaje suele ser difícil tanto para el paciente y su entorno familiar, el tratamiento debe basarse en condiciones en las que el paciente se sienta seguro, haciendo hincapié en los riesgos que conlleva presentar episodios de epilepsia y convulsiones, los cuales pueden ser persistentes y por lo tanto el especialista debe ser lo bastante cuidadoso al momento de manejar estos episodios.
Además, como se mencionó anteriormente, existen ciertos fármacos que pueden causar efectos adversos en el paciente, como el glaucoma bilateral, por lo que deberían considerarse medidas más invasivas, pero a la larga pueden representar una mejoría importante en el paciente. El factor social y psicológico debe abordarse de manera cuidadosa tanto para el paciente como para su entorno, recordando que en sus primeros años el paciente no puede entender lo que sucede a su alrededor.
CONCLUSIÓN
Es recomendable una comunicación médico-paciente-familiar, abordando el tema de desarrollo y aprendizaje que tiende a ser un poco más lento que el de un niño sin la manifestación de esta enfermedad y que al parecer el tema de imagen tiende a alejar a los pacientes de su entorno, representando un reto social del paciente y de sus padres, en los cuales los especialistas deben abordar de manera completa e interdisciplinaria para un desarrollo completo del ser humano frente a este padecimiento.
Además de que el médico de primer contacto debe establecer estrategias finas de diagnóstico para la detección de esta entidad en el paciente. Agregando el rol del especialista en psicología, dermatología y neurología para el diagnóstico, tratamiento y seguimiento del paciente a corto, mediano y largo plazo, manejando las herramientas educativas, formativas del paciente y atención especializada al trastorno de hiperactividad derivado de este padecimiento.
* Universidad Anáhuac Mayab.
** Universidad Autónoma de Yucatán.
***Instituto Dermatológico de Jalisco
Contacto: axelgb1@hotmail.com
REFERENCIAS
Akhter-Kainat, B., Sarwat-Salim, M., Sharon-Fekrat, M., et al. (2014). Sturge-Weber Syndrome and Secondary Glaucoma. EyeNet Magazine. American Academy of Ophtalmology. United States. Disponible en: https://www.aao.org/eyenet/article/sturgeweber-syndrome-secondary-glaucoma
Cerisola-Cardoso, A., Bianchi-Novoa, M., Delucchi-Botaro, G., et al. (2008). Síndrome de Sturge Weber sin presencia de angioma facial. Montevideo, Uruguay. Revista Scielo Uruguay. Disponible en: http://www.scielo.edu.uy/pdf/adp/v79n2/v79n2a07.pdf
Fernández-Concepción, O., Gómez-García, A., y Hernández-Sardiñaz, N. (1999). Síndrome de Sturge Weber. Revisión. La Habana, Cuba. Revista Scielo. Disponible en: http://scielo.sld.cu/pdf/ped/v71n3/ped05399.pdf
Fernández-Jaén, O., Sánchez-Jacob, A., y Ramos-Boludac, N. (2004). Síndrome de Sturge Weber con crisis epilépticas y calcificaciones intracraneales bilaterales en el período neonatal. Madrid, España. Revista Scielo. Disponible en: http://scielo. sld.cu/pdf/ped/v71n3/ped05399.pdf
Hernández, M., y Herrera, K. (2019). Síndrome neurocutáneo: Sturge Weber. Medigraphic. México. Disponible en: https://www.cronicascientificas.com/index.php/ediciones/edicion-xii-mayo-agosto-2019/26-ediciones/240-sindrome-neurocutaneo-sturge-weber
Higueros, E., Roe, E., Granell, E., et al. (2017). Actas dermo-si- filiográficas. Disponible en: https://www.actasdermo.org/es-sin- drome-sturge-weber-revision-articulo-S0001731016304422
Maraña-Pérez, A., Ruiz-Falcó Rojas, M., Puertas-Martín, V., et al. (2016). Análisis del Síndrome de Sturge-Weber: estudio retrospectivo de múltiples variables asociadas. Revista de Neurología. Sociedad Española de Neurología. Disponible en: https://www.elsevier.es/es-revista-neurologia-295-articulo-analisis-del-sindrome-sturge-weber-estudio-S0213485316000244
Martínez-Gutiérrez, J., López-Lancho, R., y Pérez-Blázquez, E. (2008). Síndrome de Sturge Weber: combinación de lesiones angiomatosas coroideas y orbitarias en un paciente. Revista Scielo España. Disponible en: http://scielo.isciii.es/pdf/aseo/v83n7/comunicacion1.pdf
McBride, M. (2017). Síndrome de Sturge Weber. Manual MSD. Versión para profesionales. Northeast Ohio Medical University. Ohio, Estados Unidos. Disponible en: https://www.msdmanuals.com/es-mx/profession-al/pediatr%C3%ADa/s%C3%ADndromes-neuro- cut%C3%A1neos/s%C3%ADndrome-de-sturge-weber Morales-Querol, M., Sierra-Benítez, M., y Márquez-Oquendo, J. (2017). Angiomatosis encefalotrigeminal o Síndrome de Sturge Weber. A propósito de un caso. La Habana, Cuba. Revista Médica Electrónica. Disponible en: https://www.medigraphic.com/pdfs/revmedele/me-2017/me173r.pdf
Núñez-Gamboa, E. (2018). Síndrome de Sturge Weber. Madrid, España. Portal de información de enfermedades raras y medicamentos huérfanos. Disponible en: https://www.orpha. net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=3205
Ríos, M., Barbot, C., y Pinto, P. (2012). Síndrome de Sturge Weber: variabilidad clínica y de neuroimagen. Servicio de Neuropediatría. Centro Hospitalario de Oporto. Oporto, Portugal. Disponible en: https://www.analesdepediatria.org/es-sindrome-sturge-weber-variabilidad-clinica-neuroimagen-articulo-S1695403312001543
Rodríguez-Osorio, X., López-González, F., Eiris-Puñal, J., et al. (2018). Hemisferectomía funcional: seguimiento a largo plazo en una serie de cinco casos. Revista de Neurología. Universidad de la Rioja. Disponible en: https://www.neurologia.com/articulo/2016307
Samra, J., y Portillo, G. (2004). Enfermedad de Sturge Weber. Presentación de caso y revisión del tema: Diagnóstico y tratamiento basado en medicina de evidencia. Honduras Pediátrica. Disponible en: http://www.bvs.hn/RHP/pdf/2014/pdf/Vol24-2-2014-6.pdf
Steve-Roach, E. (2020). Types of Sturge Weber Syndrome. The Sturge Weber Foundation. Texas, Estados Unidos. Disponible en: https://sturge-weber.org/new-to-swf/types-of-sturge-weber-syndrome.html