Matemáticas aplicadas para enfrentar retos biológicos: la propuesta interdisciplinaria de Ernesto Pérez Rueda

María Josefa Santos-Corral*
CIENCIA UANL / AÑO 27, No.123, enero-febrero 2024
Ernesto Pérez Rueda tiene una licenciatura en Biología por la Facultad de Ciencias-UNAM; es maestro en Investigación Biomédica Básica y doctor en Ciencias Biomédicas por el Centro de Investigación sobre Fijación de Nitrógeno-UNAM. Su área de especialidad es la Bioinformática, en particular se ha ocupado de la comparación y reconocimiento de patrones para identificar elementos proteicos que definen el destino de la expresión genética en microorganismos y, adicionalmente, junto con su equipo, ha desarrollado un enfoque novedoso en la comparación de vías metabólicas, lo cual les permitió colaborar con grupos internacionales. Sobre estos temas ha publicado más de 90 artículos científicos y dirigido tesis de licenciatura, maestría y doctorado. En 2019 fue distinguido como Research Fellow del Centro de Genómica y Bioinformática de la Universidad Mayor, en Chile. Actualmente es investigador titular C de la Unidad Mérida del Instituto de Investigaciones en Matemáticas Aplicadas y en Sistemas (IIMAS) de la UNAM.
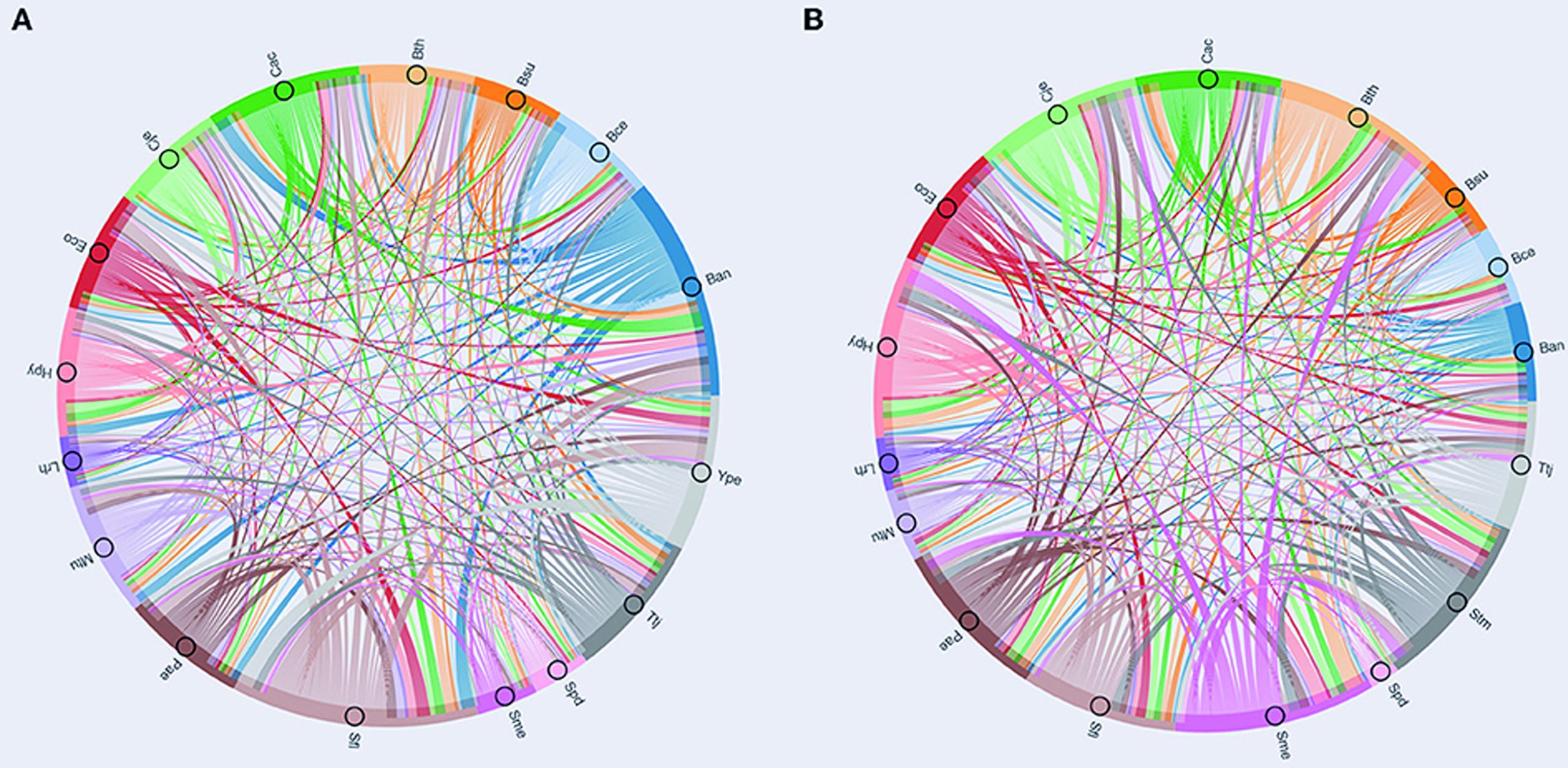
Similitud de organismos. Los colores muestran la similitud que existe en el metabolismo y la regulación genética entre las diferentes bacterias que se analizan (Galán-Vásquez y Pérez-Rueda, 2019).


¿Cómo descubre su vocación?
Es una pregunta realmente difícil. Cuando cursaba la Facultad de Ciencias, en Ciudad Universitaria, me llamaban la atención muchísimos temas, de hecho, había uno que me encantaba: los dinosaurios. Tenía un profesor de Paleontología que estudiaba fósiles de estos animales, él me acercó a la investigación. Luego tuve otras influencias, entre ellas la de un catedrático yucateco quien nos daba Bioquímica y nos ponía a pensar en asuntos como la manera en que podríamos llegar a los resultados de las reacciones y qué posibilidades había de obtener el mismo producto.
Eso despertó mi curiosidad. Además, pasé año y medio en el Instituto de Investigaciones Biomédicas, trabajando con el mal de Chagas, lo que me acercó a entender la enfermedad y a tener mi primer contacto directo con el sector que me interesó. Lo que no me gustó fue vivir en la Ciudad de México. Siempre quise irme. Así, en 1993, cuando encontré la oportunidad de hacer un posgrado en Cuernavaca, la tomé. Ahí conocí al Dr. Julio Collado Vides, mi mentor de licenciatura, maestría y doctorado, él me involucró directamente con la investigación y su proceso.
Julio era un científico que abordaba los problemas desde una perspectiva amplia, había estudiado en Estados Unidos y buscaba nuevas reglas para entender la Biología. Hacia mediados de los años noventa empezamos a recopilar cientos de datos provenientes de diversas fuentes. Lo anterior fue el detonante que despertó en mí preguntas del tipo ¿qué puedo hacer en investigación?, ¿cómo? y, específicamente ¿cómo puedo entender la naturaleza a través de lo que hago? La primera respuesta que encontré fue que una manera de entender la naturaleza es siguiendo la tendencia humana de clasificar. Así les explico a los alumnos: podemos ordenar en categorías animales, plantas, etcétera, y con ello vamos entendiendo los procesos biológicos. Con estas preguntas, y sobre todo bajo la guía de mi mentor, surgió mi vocación.

¿Cómo descubre su vocación?
Es una pregunta realmente difícil. Cuando cursaba la Facultad de Ciencias, en Ciudad Universitaria, me llamaban la atención muchísimos temas, de hecho, había uno que me encantaba: los dinosaurios. Tenía un profesor de Paleontología que estudiaba fósiles de estos animales, él me acercó a la investigación. Luego tuve otras influencias, entre ellas la de un catedrático yucateco quien nos daba Bioquímica y nos ponía a pensar en asuntos como la manera en que podríamos llegar a los resultados de las reacciones y qué posibilidades había de obtener el mismo producto.
Eso despertó mi curiosidad. Además, pasé año y medio en el Instituto de Investigaciones Biomédicas, trabajando con el mal de Chagas, lo que me acercó a entender la enfermedad y a tener mi primer contacto directo con el sector que me interesó. Lo que no me gustó fue vivir en la Ciudad de México. Siempre quise irme. Así, en 1993, cuando encontré la oportunidad de hacer un posgrado en Cuernavaca, la tomé. Ahí conocí al Dr. Julio Collado Vides, mi mentor de licenciatura, maestría y doctorado, él me involucró directamente con la investigación y su proceso.
Julio era un científico que abordaba los problemas desde una perspectiva amplia, había estudiado en Estados Unidos y buscaba nuevas reglas para entender la Biología. Hacia mediados de los años noventa empezamos a recopilar cientos de datos provenientes de diversas fuentes. Lo anterior fue el detonante que despertó en mí preguntas del tipo ¿qué puedo hacer en investigación?, ¿cómo? y, específicamente ¿cómo puedo entender la naturaleza a través de lo que hago? La primera respuesta que encontré fue que una manera de entender la naturaleza es siguiendo la tendencia humana de clasificar. Así les explico a los alumnos: podemos ordenar en categorías animales, plantas, etcétera, y con ello vamos entendiendo los procesos biológicos. Con estas preguntas, y sobre todo bajo la guía de mi mentor, surgió mi vocación.
¿Qué le hace transitar de la Biología
a la Bioinformática?
Creo que mi carrera ha sido en buena medida consecuencia no sé si del azar o de la búsqueda de preguntas novedosas. Cuando llegué a Cuernavaca, mi mentor trabajaba en gramática generativa. Mi idea en ese momento era ocuparme en evolución. Teníamos en común que no hacíamos experimentos. Yo era muy malo, no me salían las cosas; él realizaba trabajos teóricos. Empezamos en lo que se había caracterizado como Biología computacional. Con el pasar del tiempo incursionamos en la Bioinformática, una combinación entre las ciencias computacionales y la Biología, que principalmente se aplica a la Biología molecular.
En aquel entonces teníamos diversos datos en la computadora y decíamos, ¿y ahora qué hacemos?, ¿qué nos preguntamos? La respuesta la encontramos en dos vías: una manual, como lo había hecho él en su posdoctorado, o bien podíamos introducir conceptos informáticos. Ahí comenzamos a realizar trabajos en Bioinformática. Hablamos de 1994 o 1995, aproximadamente. Me gustó porque tienes que interactuar con computólogos, matemáticos y físicos. Lo interesante es que con esa mezcla llegas a soluciones que a veces no son las esperadas. Así es la Biología. Lo que sigue es explicar, desde el punto de vista biológico, qué está ocurriendo, y eso te lleva a incursionar en otros temas; además precisas valerte de conceptos de campos disciplinares distintos: Estadística, Física o Matemáticas.
Un ejemplo de lo anterior ocurrió cuando, en 1996, aproximadamente a la mitad de mi doctorado, comenzó el boom de las ciencias genómicas. El preámbulo en el montaje de toda la tecnología y secuenciar el genoma humano. En 1995 aparece el primer organismo completamente secuenciado, la bacteria Haemophilus influenzae, que puede causar neumonía. Estábamos muy contentos porque teníamos el genoma completo de una bacteria, que aunque chiquitita, marca la carrera de toda la secuenciación de organismos que se han ido liberando. A la fecha debe haber cerca de 100,000 organismos completamente secuenciados. En un lapso de 25 años pasamos de uno hasta 100,000, aproximadamente.
Esto significa que en los noventa hacíamos la comparación de un organismo contra otro, ahora la tenemos que hacer de 100,000 contra 100,000. Lo que dificulta también encontrar y “limpiar” las repeticiones. ¿Cómo quitamos ahora toda esa repetición, ese fenómeno recurrente en las grandes bases de datos? Primero hay que estar al pendiente de todos los nuevos organismos secuenciados y, segundo, de las diferentes herramientas que ayudan con el análisis de grandes cantidades de información.
Al dimensionar el problema, puedo señalar el caso de la mejor base de datos que conozco, una relacionada con la estructura de proteínas (el Protein Data Bank), tiene alrededor de 135,000 registros, los cuales puedes reducir a unos 50,000, o tal vez menos porque muchas son repeticiones. Ahora hay índices que tienen millones de secuencias, el desafío es eliminar redundancia y, posteriormente, analizarlas.
¿Qué retos supone trabajar desde la Biología con Matemáticas, integrar equipos, definir campos multidisciplinarios, abordajes, etcétera?
Eso sí nos ha costado. Sin embargo, hay ejemplos de colaboración bien interesantes, uno es el de un diseñador de algoritmos para hacer la comparación de proteínas, era físico, y básicamente la mayor parte de las notaciones que se han utilizado han sido diseñadas por físicos, quienes ven la oportunidad de analizar las proteínas como símbolos susceptibles de comparación. Ellos realizan los avances más significativos. En mi caso ha sido un poco difícil la interacción con gente de otra área; sobre todo explicar el fenómeno que queremos explorar.
Tal vez el éxito más sobresaliente que hemos tenido es con una colega del IIMAS, en Ciudad Universitaria, quien labora con algoritmos genéticos y fue mi entrada al Instituto. Ha sido difícil hablar o explicar los fenómenos que nos interesan a los matemáticos. Hemos tenido poca interacción. Hay una cierta barrera, no digo con todos, pero sí para que los biólogos sean aceptados en Matemáticas y los matemáticos en Biología. Se siguen viendo separadas, creo que nos falta ampliar nuestra perspectiva e integrarnos mejor.
¿En qué aporta a sus investigaciones el estar en un centro de Matemáticas aplicadas?
Me ha ayudado. Aunque estoy en el IIMAS-Mérida, considero que me pierdo de las cosas que ocurren en Ciudad Universitaria, donde se encuentra lo fuerte del Instituto, así que me he tenido que ir adaptando a lo que tenemos en Yucatán, lo que no es nuevo, pues también lo hice cuando me encontraba en Cuernavaca, primero en el Centro de Ciencias Genómicas y después en el Instituto de Biotecnología; en ambos casos mi área de interacción era principalmente con biólogos, aunque hablaba con algunos físicos que se ubicaban el Instituto de Física.
La relación no era muy buena, sobre todo porque a veces no lograba explicar la idea o ellos no se mostraban muy interesados en lo que hacíamos. Y ahora, en el Instituto, tengo dos colaboradores muy cercanos, uno que se especializa en algoritmos genéticos y otro en teoría de redes. Entonces, tengo personal muy cercano que me ayuda a resolver los problemas, sobre todo de la parte de diseño de algoritmos.
¿Qué aplicaciones han tenido sus trabajos académicos?
El grupo de bioinformática y genómica comparativa en el que colaboro codifica genes de ciertos organismos, para después tomarlos de referencia en la anotación de otros. A partir de ello puede caracterizar y buscar propiedades de éstos; verbigracia, las enzimas que degradan el plástico se caracterizan en un organismo y luego se pueden buscar en otros, utilizando algoritmos de comparación de patrones.
Este equipo está dividido en dos grandes líneas: la primera trata de entender cómo se regulan los genes en los organismos, sobre todo en bacterias. En la segunda comparamos y analizamos las vías metabólicas de los organismos. En ambas hemos tenido avances significativos. En específico, al observar el repertorio de proteínas que regulan la especie genética en un organismo moderno. El trabajo pionero (publicado en 1998) fue sobre Escherichia coli.
En el caso del metabolismo, nuestro grupo ha logrado obtener dos cosas: la perspectiva meramente teórica, la parte filosófica o evolutiva; la otra, en la que nos desempeñamos actualmente, tiene una aplicación más biotecnológica: mostrar que hay rutas alternativas a las vías metabólicas principales. Esto significa que, si bien hay una vía principal, que suele ser la que más se ha estudiado, hay alternativas que incluso pueden estar más conservadas. Con ello podemos trazar nuevos caminos en la producción de un compuesto parecido, tal vez, con mayor eficiencia.
Si lo aterrizas a la Biotecnología, significa la construcción de compuestos por rutas menos costosas. Adicionalmente, hemos trabajado en colaboración con gente de Yucatán, en el dise- ño de vacunas. Con ellos analizamos algunas proteínas del virus chikungunya, un problema muy serio en la península. Identificamos por aquellas regiones las proteínas de este virus con una alta capacidad inmunogénica. Utilizamos herramientas computacionales y las contrastamos con la información reportada en la bibliografía. Ahora mismo esperamos los aná- lisis experimentales.
También, con colegas de Ensenada, llevamos tiempo ocupándonos en una proteína que se utiliza en el combate de leucemia infantil. En principio se podría obtener de una manera más fácil y barata. Se identifican las regiones inmunogénicas, se enmascaran y que no desencadenen la respuesta inmune en el humano, pero sí dejarlas directamente sobre las células cancerosas. La idea es que este vehículo llegue a ellas envuelto en una carcasa, se abra y libere la proteína, a manera de caballo de Troya.
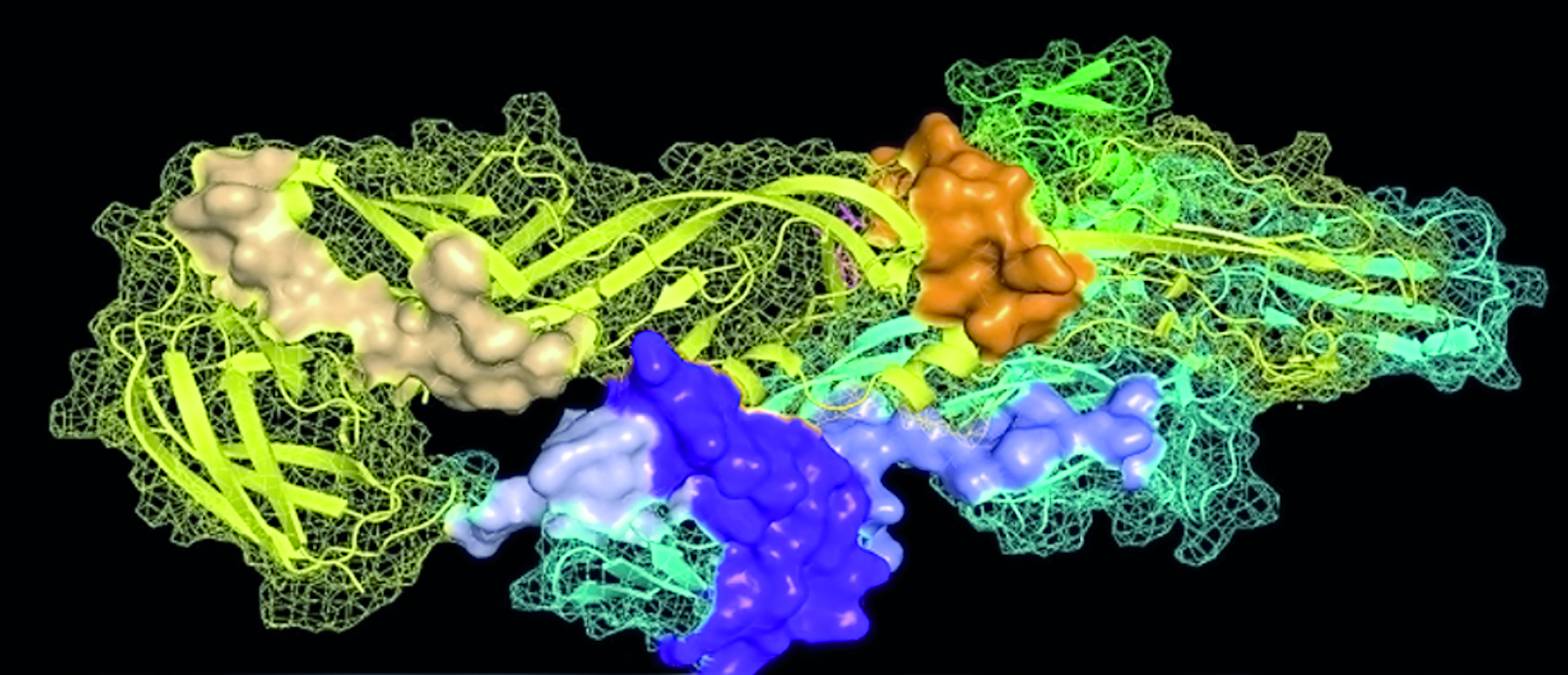
Virus chikunguya. Los colores muestran las regiones con potencial antigénico para la respuesta inmune (Sánchez-Burgos et al., 2021).
¿Dónde considera que han estado sus aportes más importantes:Biología, Matemáticas, un nuevo campo disciplinar?…
Creo que en Biología. En este momento estoy colaborando con un estudiante de doctorado en el diseño de técnicas de machine learning en el análisis de secuencias de DNA, una especie de clasificador. Allí la aportación va en dos sentidos: uno, el algoritmo tal cual para clasificar; utilizamos las propiedades biofísicas de los nucleótidos y convertirlos en atributos numéricos. Vemos una cadena de ADN como si fueran atributos numéricos y los comparamos, esto nos lleva a encontrar una región importante con una función determinada. Ese es el aporte en la parte biológica, es decir, vamos a ver cuáles son las regiones que identificamos sin considerar la secuencia.
El segundo aporte es en la parte informática, el diseño del algoritmo que toma en cuenta datos biofísicos. Ahí convergen varias aristas: Biofísica, Física y cómputo, donde tenemos que llevar a cabo el diseño de un algoritmo que nos analiza la secuencia. Ejemplo de lo anterior es lo que hicimos con Edgardo Galán, del IIMAS; analizamos, desde la teoría de redes, la interacción de los distintos fármacos aprobados por la Federal Drug Agency (FDA) de Estados Unidos con proteínas y genes. Encontramos que un mismo fármaco puede reconocer distintas cosas.
Hay medicamentos agresivos recetados en el tratamiento de Alzheimer, o esquizofrenia, con más efectos secundarios que beneficios. Para ilustrar mencionaré la amoxapine, prescrita como antidepresivo, reconoce los mismos blancos que otros 380 fármacos; sin embargo, los otros blancos que reconoce se asocian a medicamentos que tratan alcoholismo, alergias o hiperplasia prostática. De ahí derivan montones de situaciones, una de ellas el reposicionamiento de medicinas, básicamente lo que hicieron cuando la pandemia de COVID. Si bien no inventamos nada nuevo, encontramos cuál es la diversidad de drogas que reconocen todas las diferentes proteínas y cuáles son reconocidas por la mayor canti- dad de fármacos. Hay cosas muy interesantes y que podrían ser preocupantes, por ejemplo, existe un gen reconocido hasta por 15 medicamentos contra 15 enfermedades.
En esa misma línea trabajamos con un colega del IPN, analizando los genes que se expresan en diversas condiciones de cáncer de mama. Los identificamos utilizando una metodología de redes, para saber cuáles están más conectados entre las distintas condiciones. Lo que sigue es encontrar los fármacos reconocidos por estos genes sobreexpresados. Posteriormente queremos, desde el campo del docking, descubrir nuevas interacciones entre medicamentos y proteínas.

¿Qué le ha dado la UNAM al doctor Pérez Rueda y usted qué le ha dado a la institución?
Yo te puedo decir que la UNAM me ha dado todo. Me ha permitido crecer como investigador, como persona. Soy universitario desde 1986, cuando ingresé al CCH Sur, a los 14 años, todavía recuerdo mi número de matrícula. La UNAM me ha permitido crecer y me ha otorgado libertad de indagar lo que considero interesante.
Pienso que le he regresado mucho menos de lo que ella me ha dado, y citaría que he tratado de devolver lo recibido formando estudiantes, lo más importante del quehacer de la UNAM, o al menos mi labor más importante. Me gusta muchísimo la investigación, me apasiona. Diría Dobzhansky: «Nada tiene sentido en Biología si no está en la luz de la revolución». Yo diría, retomándolo: la investigación no tiene ningún sentido si no es a la luz de la formación de estudiantes. Si no formas estudiantes, no hay sentido. Algo que siempre les digo a los jóvenes, cuando ingresan, es que la única diferencia entre ellos y yo es el tiempo que tenemos dedicados al tema, al área. Los veo como colegas en ciernes, simplemente porque están empezando.
*Universidad Nacional Autónoma de México, Ciudad de México, México.
Contacto: mjsantos@sociales.unam.mx
REFERENCIAS
Galán-Vásquez, E., y Pérez-Rueda, E. (2019). Identification of Modules With Similar Gene Regulation and Metabolic Functions Based on Co-expression Data. Front. Mol. Biosci. 6:139. https://doi.org/10.3389/fmolb.2019.00139
Martínez, G.S., Pérez-Rueda, E., Sarkar, S., et al. (2022). Machine learning and statistics shape a novel path in archaeal promoter annotation. BMC Bioinformatics. 23:171. https://doi.org/10.1186/s12859-022-04714-x
Sánchez-Burgos, G.G., et al. (2021). In Silico Identification of Chikungunya Virus Band T-Cell Epitopes with High Antigenic Potential for Vaccine Development. Viruses. 13:2360. https://doi.org/10.3390/v13122360